











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Many molecules with some type of biological activity possess one or more stereogenic centers. When these molecules are found in very small amount, it is necessary to synthesize them in the laboratory to confirm their biological activities and to prepare analogues to study them further. It is important to develop asymmetric methods that are practical, inexpensive and efficient to obtain these molecules with exquisite control of their stereochemistry. There are several strategies to control the stereochemistry, each one with its own advantages and disadvantages. Among these, we can find resolution of a racemic mixture using a homochiral resolution agent to obtain a diastereomeric mixture, which is separated using physical methods to release the original enantiomers. Another strategy is the use of natural aminoacids, saccharides, alkaloids, etc. from the chiral pool to build the desired target molecule. In asymmetric catalysis, a chiral catalyst or ligand is employed to selectively create chirality in an achiral substrate. In the chiral auxiliary strategy, a non-chiral substrate A is covalently attached to the chiral auxiliary Xc to give an adduct Xc-A, Figure 1. The adduct Xc-A provides a predictable stereo-differentiated environment on a prochiral atom during the chemical reaction to be transformed diastereoselectively Xc-A*. Finally, the diastereomer Xc-A* obtained is cleaved to recover the chiral auxiliary Xc and release the chiral molecule A*. One of the advantages of this method is its reliability. An obvious disadvantage is the increase in the number of steps during the synthesis.
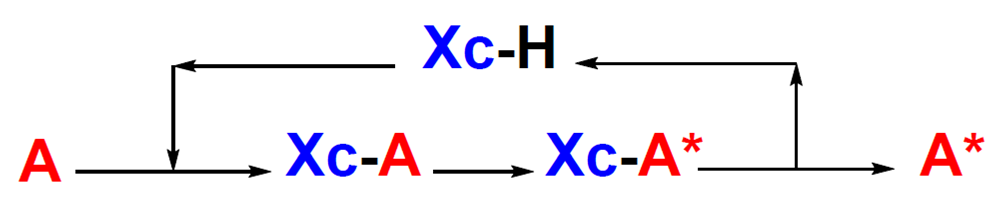
There is a large number of applications employing chiral auxiliaries. Several chiral molecules have been used in this strategy. Arguably, the most well-known chiral auxiliaries are Evans’ 1,3-oxazolidin-2-ones [1,2] in the synthesis of natural products [3]. Amino acid-based Evans' oxazolidinones have been widely used in the synthesis of natural products and compounds with pharmacological activity [4]. However, in the last two decades the popularity in asymmetric synthesis of the sulfur analogs 1,3-oxazolidine-2-thiones and 1,3-thiazolidine-2-thiones has risen because in many cases they present excellent effectiveness and convenience as chiral auxiliaries[5,6]. These sulfur based chiral auxiliaries, similarly to Evans’ auxiliaries, can be rapidly obtained from amino acids using conventional and non-conventional methods. [7-10]
Aldol reactions
Chiral auxiliaries are commonly employed to create carbon-carbon bonds in alkylations [11], aldol reactions [12-14], and others [15,16]. Evans’ oxazolidinones deliver aldol products with outstanding diastereoselectivities when the N-acyl group on the oxazolidinone is a propionyl group 1 (Scheme 1). A Z-enolate is formed in the presence of the boron catalyst, and the “Evans” syn-aldol product 2 is delivered.[17] Interestingly, by adding another equivalent of Lewis acid, the “non-Evans” syn-product 3 can now be obtained.[18] Unfortunately, when the N-acyl on the oxazolidinone is an acetyl group, no diastereoselectivity is observed in this reaction.[19]

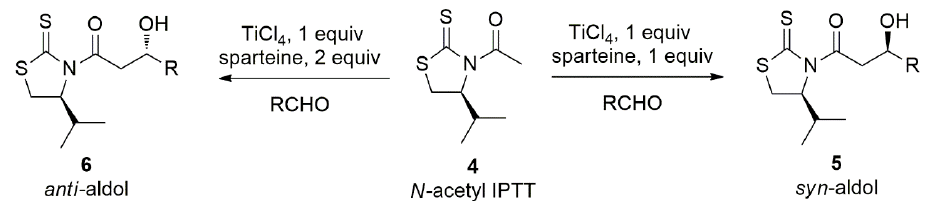
In contrast to oxazolidinone chiral auxiliaries, sulfur-based analogs have proved to be very effective in aldol type reactions when the N-acyl group is an acetyl. The aldol condensation with the acetyl group was initially reported by Nagao.[20,21] Nagao utilized tin(II) as the Lewis acid and obtained the syn-acetyl aldol product with high diastereoselectivity. We studied the aldol reaction with N-acetyl 4(S)-isopropylthiazolidinethione (4), and found that utilizing one equiv. of TiCl4 and one equiv. of sparteine, the syn-acetyl product 5 was obtained with high diasteroselectivity (Scheme 2). Increasing the amount of Lewis acid did not change the outcome of the reaction. It is preferred to utilize a solution of TiCl4 because it is easier to handle than the tin(II) Lewis acid.[22] When we used only one equiv. of TiCl4 and now two equiv. of sparteine, in most cases, it was noticed a reverse in the diastereoselectivity, but dr depended on the nature of the aldehyde employed. The acetate syn product 5 was explained by a closed transition state where the titanium is coordinated to both oxygens and the thiocarbonyl sulfur atom. On the other hand, the anti-product 6 was explained by an open transition state where the titanium is bound only to two oxygens but not to the sulfur atom allowing a more stable anti conformation between carbonyl and thiocarbonyl groups [23].
This acetate aldol methodology was applied to the construction of the C10-C17 fragment 8 of cytotoxic aurisides and callipeltosides (Scheme 3). [24] Aurisides A and B are brominated and glycosylated 14-membered macrolides isolated from the sea hare Dolabella auricularia by Yamada’s group in 1996.[25] Callipeltosides A and B are similar glycosylated 14-membered macrolides isolated from the marine sponge Callipelta sp. by Minale’s group in the same year.[26] Thus, 5-bromopentadienal was prepared from pyridinium sulfonate in two steps.[27] The aldehyde was reacted with N-acetyl 4(S)-IPTT 4 and one equiv. of TiCl4 and Hunig’s base to give the desired syn-aldol product 7 in 75% yield. The aldol product 7 was silylated and reduced with DIBAL-H to obtain directly the corresponding aldehyde, without the need to prepare a Weinreb amide, which was then transformed into the C10-C17 side chain 8 of aurisides. This was an important fragment in order to study the ring construction of the 12-membered macrolactone.[28]

Resolution of a racemic mixture of acids
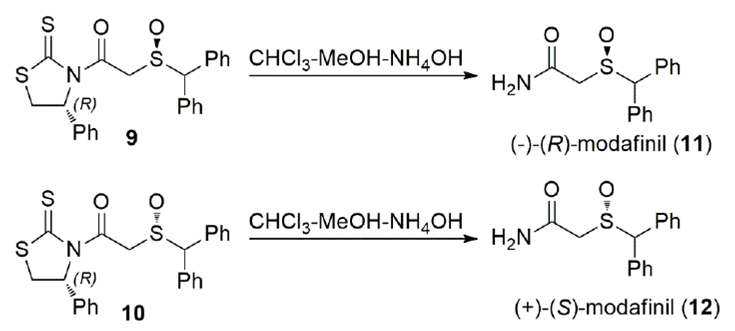
Modafinil (11/12) is a unique CNS stimulant devoid of addiction liability and it is used clinically to treat narcolepsy (Scheme 4).[29] Modafinil is a rather small molecule with a sulfoxide stereogenic center. We decided to use a chiral auxiliary to separate the diastereomeric mixture of thiazolidinethiones and obtain optically pure compounds which could be easily transformed into each enantiomer of modafinil.[30] Thus, modafinic acid was attached to the 4(R)-phenylthiazolidinethione auxiliary and the mixture of diastereomers 9 and 10 was separated by preparative chromatography. Treatment of the thiazolidinethione imides with ammonium hydroxide allowed us to obtain directly optically pure enantiomers 11 and 12 of modafinil in very high yield and the chiral auxiliary was recovered.
Addition on iminium ions and natural products synthesis
(-)-Stemoamide is a tricyclic alkaloid with four contiguous stereogenic centers found in Stemona tuberosa.[31] The synthesis of this alkaloid has been carried out several times to showcase new synthetic methodology. We envisioned a synthetic strategy where we could control the four stereocenters of (-)-stemoamide with the aid of a chiral auxiliary.[32] The addition of chlorotitanium enolates of N-acetyl IPTT 4 to five-membered N-acyl iminium ions was investigated (Scheme 5).[33] The anti-addition product 14 was selectively obtained when two equiv. of TiCl4 and 1.2 equiv. of Hunig's base were employed in the addition of N-acetyl IPTT 4 to N-alkyl 5-acetoxypyrrolidinone 13.
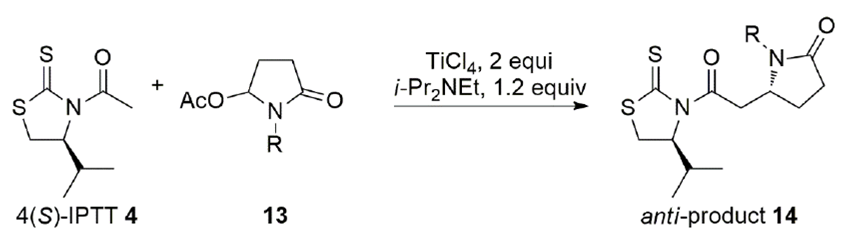
We employed the 4(R)-PhTT for the synthesis of (-)-stemoamide (17) because the two isomers of PhTT are easily obtained from the corresponding enantiomer of phenylglycine and are inexpensive (Scheme 6). Evans had reported recently a novel strategy to obtain the anti-aldol product utilizing these sulfur-based chiral auxiliaries.[34,35] This methodology is only applicable to non-enolizable aldehydes. Thus, the iminium addition product 15 was reacted with cinnamaldehyde utilizing trimethylsilyl chloride, trimethylamine and cat. amount of MgBr2∙OEt2. The aldol product was then protected as the silyl ether 16. Thiazolidinethione 16 was reduced directly into the corresponding aldehyde in low yield. Excellent yields were obtained when the aldehyde was obtained in two steps (reduction/oxidation). Wittig olefination delivered a methyl vinyl ether. The lactone ring was then constructed via a methyl vinyl ether hydrolysis and cyclization with the free alcohol. The azepane ring was constructed via ruthenium-catalyzed ring closing metathesis. X-Ray analysis of synthetic (-)-stemoamide was obtained to confirm the stereochemistry of the four contiguous stereocenters.

Michael additions
Erogorgiane is a highly lipophilic diterpene isolated from the West Indian gorgonian octocoral Pseudoterogorgia elisabethae.[36] Erogorgiane and its 7-hydroxy analogue have showed potent anti-tuberculosis activity. Interestingly, erogorgiane was also isolated from the root bark extract of Leucophyllum frutescens by Waksman (Universidad Autónoma de Nuevo Leon).[37] We proposed a synthetic strategy for the synthesis of erogorgiane where the stereochemistry of C-1 and C-11 could be controlled by Michael addition of a methylcuprate nucleophile to an α,β-unsaturated sulfur-based imide chiral auxiliary (Scheme 7). In collaboration with Benemérita Universidad Autónoma de Puebla, we reported that addition of methylcuprates, generated in situ using an excess of a 1:2 mixture of CuI-DMS and the Grignard reagent to N-enoyl oxazolidinethione 18 in the presence of excess TMSI gave preferentially the anti-diastereomer 19.[38] This Michael addition took place when the conformation of the substrate was syn-s-cis.

One of the advantages of using this type of sulfur-based chiral auxiliaries is that they can be easily displaced with carbon nucleophiles.[39] The β-keto ester 20 was obtained by malonate displacement/decarboxylation of the oxazolidinethione in imide 19 utilizing potassium salt of monoethyl malonate and MgCl2 in the presence of imidazole (Scheme 7).[40] The corresponding (-diazo-(-ketoester 21 was prepared from the (-ketoester 20 and underwent an aromatic C-H insertion in the presence of Rh(II) catalyst.
Sulfur-based chiral auxiliaries 22 can also act as nucleophiles in Michael additions to N-enoyl imides 23 (Scheme 8).[41] This conjugate addition was observed in the presence of excess triethylamine in dichloromethane at reflux. The addition takes place by the nitrogen of the heterocycle with high diastereoselectivities. This reaction occurs on the anti-s-cis conformation of the N-enoyl sulfur-based auxiliary 23 to give Michael addition product 24.
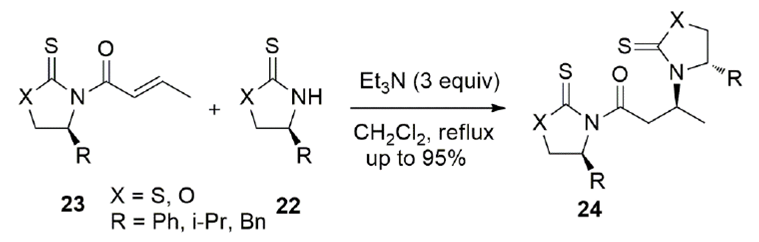
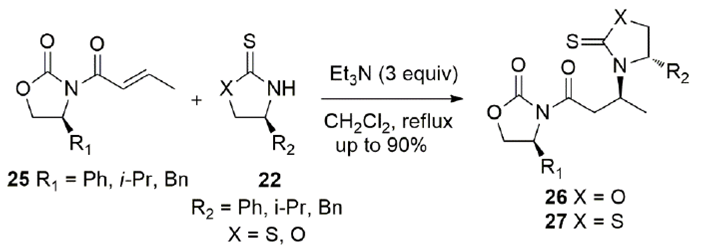
A complex mixture is observed when a thiazolidinethione is added to N-crotonyl oxazolidinethione or oxazolidinethione is added to N-crotonyl thiazolidinethione because a concomitant trans-acylation occurs.[42] However, when the Michael acceptor is N-crotonyl oxazolidinone 25 and the nucleophiles are thiazolidinethione (X = S) or oxazolidinethione (X = O) 22, no trans-acylation is observed and Michael addition products 26 and 27 are cleanly obtained (Scheme 9).
Cascade reactions
N-Enoyl oxazolidinethiones and thiazolidinethiones can undergo an intramolecular conjugate addition of the sulfur atom in the presence of Lewis or Bronsted acid.[43-46] Although, the preferred conformation of this type of compounds is the anti-s-cis in solid state, the anti-s-trans conformation 28 is required for the intramolecular conjugate addition. Based on this precedent, we designed a cascade reaction by the addition of an aldehyde in the presence of TiCl4 or BF3∙OEt2.[47] Unique cage-type compounds 29 and 30 possessing a quaternary carbon attached to four heteroatoms were prepared in diastereoselective manner (Scheme 10).

Five-member heterocyclic chiral auxiliaries can be easily identified by their characteristic carbonyl or thiocarbonyl chemical shift in 13C NMR. Having the thiocarbonyl at 200.8 ppm in thiazolidinethione 31 and at 189.5 ppm in oxazolidinethione 33 and the carbonyl at 175.8 ppm in thiazolidinone 32 and at 160.4 ppm in oxazolidinone 34 for the valine derived chiral auxiliaries (Scheme 11). Thiazolidinethiones can undergo desulfurization/oxygenation to thiazolidinones when heated in a sealed tube containing propylene oxide.[48] Similarly, oxazolidinethiones are transformed into oxazolidinones under the same reaction conditions.

Indene-based auxiliary
More sterically encumbered thiazolidinethiones and oxazolidinethiones have been prepared from non-natural aminoalcohols/aminols.[49-53] However, these new chiral auxiliaries are more expensive and laborious to obtain. For this reason, we envisioned preparing an inexpensive thiazolidinethione based on Ghosh’s indene-based oxazolidinone.[54] This sulfur-based chiral auxiliary was prepared from commercially available trans-1-amino-2-indanol.[55] This indene-based thiazolidinethione 35 was shown to be very effective in the acetate aldol reaction with propionaldehyde to give product 36 in excellent yield and very high diastereoselectivity (Scheme 12). The N-propionyl derivative 37 was shown to work very successfully in obtaining the non-Evans aldol product 38 and also the Evans-aldol product 39. The same derivative gave high diastereomeric ratio in the anti-aldol product 40 catalyzed by magnesium bromide in ethyl acetate.

The N-acetyl indene-based thiazolidinethione 35 was employed to introduce chirality twice during our synthesis of the marine macrolide aurisides (Scheme 13).[56] First, the acetate aldol reaction was carried out with 5-bromopentadienal using one equiv. of TiCl4 and one equiv. of sparteine. Reaction yield and diasteroselectivity of the aldol reaction were similar to the results using the IPTT chiral auxiliary. The aldol product 41 was reduced directly to the aldehyde 43 after silylation and it was transformed into the corresponding alkyne C10-C17 fragment 44.

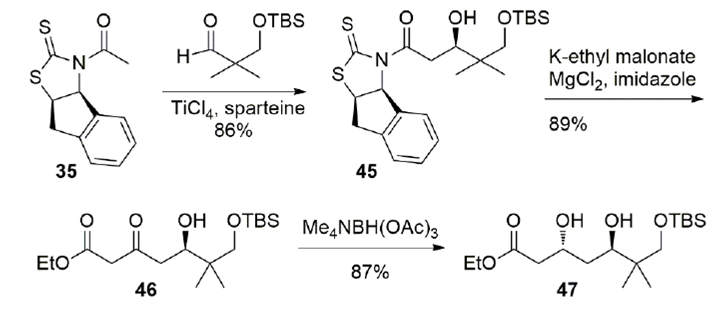
The same N-acetyl chiral auxiliary 35 was employed for the construction of the northern fragment 47 of aurisides (Scheme 14). The aldol product 45 was displaced with potassium ethyl malonate and the β-ketoester 46 subjected to hydroxyl-directed reduction with tetrabutylammonium triacetoxyborohydride to give the 1,3-anti-diol 47. This molecule was then cyclized to the lactone in acidic media. A few more steps were required to attach the two fragments and complete the synthesis of the auriside aglycone.
Another interesting application of the indene-based thiazolidinethione auxiliary is in the synthesis of simplactones A and B.[57] Simplactones A and B were isolated in trace amounts from marine sponge Plakortis simplex by Fattorusso in 1999.[58] These natural products are (-valerolactones possessing two stereogenic centers. First, an aldol reaction of N-butanoyl imide 48 was carried out with formaldehyde, in the form of trioxane to give aldol product 49 in high diastereoselectivity (Scheme 15). The reaction was successful in creating the stereochemistry of C-4 of the natural product. The aldol product 49 was silylated and directly reduced to the chiral aldehyde 51. Aldehyde 51 was subjected to an acetate aldol reaction with imide 35. Interestingly, we observed that the stereochemistry of the alcohol 52 is only influenced by the stereochemistry of the chiral auxiliary and not by the aldehyde’s chirality. The aldol product 52 was treated with acid to obtain directly the (3R,4S)-simplactone (53). In a similar fashion, the (3R,4R)-simplactone was also prepared.

Conclusion
In conclusion, in this mini-review we have shown the use of sulfur-based chiral auxiliaries in different applications for the syntheses of natural products and important medicinal agents. These chiral auxiliaries are easy to prepare, provide a high degree of diastereoselectivity, and are easy to remove. In addition, the yellow color of the N-acyl derivatives and crystalline nature, make them easy to purify. Our research not only included target-oriented syntheses, but also some interesting aspects in the chemistry of sulfur-based chiral auxiliaries. We have shown in these applications that the use of chiral auxiliaries is a very convenient strategy to obtain valuable optically pure compounds. We believe that the chemistry of these sulfur-based chiral auxiliaries will continue to show practical advantages in the syntheses of biologically relevant molecules.

