











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introduction
The Coronavirus disease 2019 (COVID-19), caused by SARS-CoV-2 infection, has spread rapidly worldwide and has become a global public health emergency (Yang et al., 2020). Coronavirus is related to severe acute respiratory syndrome (SARS), type 2 (Yoshimoto, 2020). Some of the main viral proteins of SARS-CoV-2 are the spike protein (S; Spike), the main structural protein in cell invasion mediated by ACE2 receptors, and the host cell protein TMPRSS2. Protein S is currently the most investigated therapeutic target. Another important protein is the major nonstructural protease Mpro (also known as 3CLPro) which facilitates the proteolytic processing of polyproteins (Da Silva et al., 2020; Paraiso et al., 2020). RNA-dependent RNA polymerase (RdRp) plays a crucial role in the viral cycle. RdRp is the most conserved and accessible region of RNA viruses; targeting this region for inhibition of viral replication may be a practical therapeutic approach (Aftab et al., 2020).
In addition to the development and use of vaccines, the use of pre-existing drugs has been proposed. However, the effectiveness of these is limited. The use of existing antivirals has also been reallocated to reduce time and cost compared to new drug discoveries. Some existing drugs evaluated versus SARS-CoV-2 are Remdesivir, Lopinavir/Ritonavir (Yang et al., 2020).
Effect and mechanism of action of Remdesivir
Nucleoside analogs are antiviral agents that have shown efficacy against viruses, such as coronaviruses, HIV, hepatitis B, and C (Eastman et al., 2020). Remdesivir is a nucleoside analog prodrug metabolized within cells to an active nucleoside triphosphate (NTP) metabolite (Moneriz & Castro-Salguedo, 2020). The active metabolite targets the viral RNA replication machinery and acts as a substrate for RdRp, which competes with ATP to incorporate new strands to form the RNA chain. The incorporation of remdesivir disrupts downstream molecular processes (Malin et al., 2020). The viral RdRp is the target protein of the active metabolite, although in silico studies have shown that remdesivir can also strongly bind the Mpro protein (Nguyen et al., 2020).
Bioactive Compounds
In fruits, vegetables, and plants, bioactive compounds such as vitamins, phytochemicals, and phenolic compounds (flavonoids and carotenoids) have health benefits. These compounds have biological properties such as antioxidant, anticancer, and antimicrobial activities (Torres-León et al., 2017). Antimicrobial properties include antiviral activity. Antiviral components of various fruits and plants can act on viruses and host cells to prevent viral infection (Mukhtar et al., 2008; Bright & Gilling, 2016; Ben-Shabat et al., 2019).
In silico studies
In silico studies have been investigated to evaluate the potential interaction and affinity of bioactive compounds against SARS-CoV-2. It has been reported through in silico and in vitro studies that some polyphenols have the potential to inhibit RdRP and SARS-CoV-2 viral proteases (Torres-León et al., 2020; Singh et al., 2020). Polyphenols are promising compounds for affinity with viral proteases involved in viral replication (Paraiso et al., 2020; Singh et al., 2020). In silico studies have been conducted on some medicinal plants rich in hydrolyzable tannins, which can be used to treat SARS-CoV-2 (Khalifa et al., 2020). Other studies suggest that flavonoid glycosides are inhibitors of SARS-CoV-2 in Mpro and RdRp (Da Silva et al., 2020). Therefore, evaluating the bioactive compounds through a molecular docking approach to inhibit SARS-CoV-2 proteins is very relevant (Tallei et al., 2020).
Software programs for molecular docking are commercially available. However, some are quite expensive, limiting their use, and some licenses must be renewed yearly (Benfenati et al., 2010). Some free software programs are available online, with zero costs, and can be easily used to test any chemical. These types of programs can help reduce the time in the selection of bioactive compounds and facilitate their analysis in vitro or in vivo.
In Mexico, there is a large number of medicinal plants that are used in traditional medicine. The study of these plants has contributed to the discovery of new substances with biological activity. For this reason, plants such as Parthenium argentatum A.Gray, Turnera diffusa Willd. ex Schult., Larrea tridentata (Sessé & Moc. ex DC.) Coville, Taraxacum officinale (L.) Weber ex F.H.Wigg, Moringa oleifera L., Eucalyptus camaldulensis Dehnh., and Bougainvillea glabra Choisy were selected. These plants are distributed throughout the Mexican territory and are used as medicinal plants. Molecules from each plant were selected based on previous research on the presence of viral or biological activity. The bioactive compounds against the SARS-CoV-2, Mpro (PDB; 6LU7), and RdRp (PDB; 7BV2) proteins were tested by molecular docking using two servers available online.
Material and Methods
Molecules and plants
Plants and their molecules in parentheses: Parthenium argentatum A.Gray (Neochlorogenic acid; CID: 5280633), Turnera diffusa Willd. ex Schult. (Luteolin 7-glucoside; CID: 5280637, Syringetin glucopyranoside; CID: 16109838), Larrea tridentata (Sessé & Moc. ex DC.) Coville (Nordihydroguayretic acid; CID: 4534), Moringa oleifera Lam. (Kaempferol; CID: 5280863, Apigenine 7 O-glucoside; CID: 5280704), Eucalyptus camaldulensis Dehnh. (Cypellocarpin B; CID: 10506563, Cypellocarpin C; CID: 10625791), Taraxacum officinale (L.) Weber ex F.H.Wigg. (Betulin; CID: 72326), and Bougainvillea glabra Choisy (Vitexin; CID: 5280441) were selected for this study. Remdesivir (CID: 121304016) was used as a positive control. This molecule has been approved by the FDA for use in patients with COVID-19 (FDA, 2020) and has extensive research as a possible treatment against COVID-19 (Beigel et al., 2020; Ko et al., 2020; Ma et al., 2021; Wang et al., 2020; Yang et al., 2020). 2D molecules were obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov/) in SDF format.
Determination of Lipophilicity
The SwissADME web-based tool (http://www.swissadme.ch/index.php) was used to predict lipophilicity (LogP). Lipophilicity is an important factor for absorption, distribution in the body, penetration through vital membranes and biological barriers, metabolism, and excretion of a compound (ADME properties).
The logP of a compound intended for oral administration must be <5. Therefore, the selected molecules were evaluated to determine which ones comply with this rule.
In silico molecular docking
The molecules were evaluated as potential ligands for the SARS-CoV-2 proteins, Mpro (PDB: 6LU7) and RdRp (PDB: 7BV2), using two online molecular docking servers (both servers are freely available for use). Molecules downloaded from PubChem were not modified before analysis.
Currently, there are many molecular docking servers; some need to be installed on a computer and supplemented with more programs. Also, most of these programs have to be purchased. However, there are also free programs for molecular docking. The free online programs have economic (zero costs), infrastructure (do not require high-quality computers), storage (the information generated by these programs is stored on their servers), and analysis (the proteins of interest are already available) advantages. The disadvantages are the limited options to change details and the lower availability of computational resources for high-performance virtual detection experiments. However, online servers for molecular docking are useful for searching for bioactive compounds against SARS-CoV-2.
In the present study, we use two servers available online and free. For the selection of the two servers, the BIOPEP-UWM database of the University of Mazury in Olsztyn, Poland (https://biochemia.uwm.edu.pl/en/docking-2/) was entered. The database has 23 online programs for molecular docking: B-AceP tool, AMMOS2, AutoDock Vina, CB-Dock, BINANA, ClusPro, CovalentDock Cloud, COVID-19 Docking Server, DockThor, EDock, FitDock, GalaxyPEPDOCK, HawkDock, HPEPDOCK, Hex, InstaDock, PIPER-FlexPepDock, ProteinsPlus, SwissDock, systemsDock, UNRES server, Webina, ZDOCK. Of all the programs, we selected two non-randomly: COVID-19 Docking Server (CDS) and DockThor (DT).
CDS
CDS (https://ncov.schanglab.org.cn/index.php) (Kong et al., 2020) has SARS-CoV-2 proteins available for molecular docking; this facilitates the evaluation of the affinities and binding modes between proteins and ligands (small molecules, peptides, and antibodies). The proteins used to carry out the molecular docking were Mpro and RdRp (RTP site). Molecules for docking were loaded in SDF format.
Autodock Vina is used as a docking engine in CDS. The docking box is defined as the center of the native ligand coordinate with 30 Å × 30 Å × 30 Å length to include residues from the entire cavity. CDS uses MGLTools to add hydrogens and prepare proteins and ligands. The level of exhaustiveness was the default (12). The analysis was carried out with the proteins Mpro (PDB: 6LU7) and RdRp (PDB: 7BV2). The protein of interest was selected, and the molecule to be analyzed was added. The score value of model 1 was used (in kcal/mol).
DT
DT (https://dockthor.lncc.br/v2/) (da Silveira et al., 2019; Santos et al., 2020) has SARS-CoV-2 viral proteins specific for molecular docking. Therefore, only the molecule of interest is loaded, and the protein to be evaluated is selected. The 2D structures of the molecules in SDF format were converted to PDB format with the SMILES online converter (https://cactus.nci.nih.gov/translate/). The wild-type structures Nsp5-Mpro (PDB: 6LU7) and Nsp12-RdRp (PDB:7BV2) were used for the proteins. As docking parameters, the catalytic binding site of both proteins and the grid box size were defined as 20 Å × 20 Å × 20 Å. The standard algorithm precision (i.e., 1,000,000 evaluations, a population size of 750, and 24 runs) was used.
Protein-ligand binding analysis
Results from the DT server that had similar or better results than control Remdesivir (Cypellocarpins B and C, Luteolin 7-glucoside, and Syringetin glucopyranoside) were downloaded to analyze ligand-protein binding interactions using Discovery Studio Visualizer. 2D images representing the molecule-protein interaction were generated.
Data Analysis
The analysis was performed using descriptive methods. The results correspond to the binding energy (kcal/mol) and the type of bond between the compound and the protein. Binding energy values indicate affinity between compounds and proteins. Negative binding energy means that the compound has an affinity for the protein, and a positive one shows that the compound has no affinity for the protein. The negative bond energy value indicates a spontaneous reaction and a stable system that allows bond formation.
Results and Discussion
Determination of Lipophilicity
The results showed that 10 molecules have LogP values within the range (<5), Remdesivir (1.50), Neochlorogenic acid (-0.38), Luteolin 7-glucoside (0.16), Syringetin glucopyranoside (-1.55), Nordihydroguayretic acid (3.29), Kaempferol (1.58), Apigenin 7 O-glucoside (0.55), Cypellocarpin B (0.70), Cypellocarpin C (1.04), Betulin (6.36), Vitexin (-0.07). Betulin showed a higher LogP value. This result indicates that most molecules are similar to drugs, and their study as possible treatments against SARS-CoV-2 is appropriate.
Analysis of molecular docking of bioactive molecules
Table 1 shows the docking between molecules and protein structures of SARS-CoV-2, Mpro, and RdRp. These scores represent the binding energy (kcal/mol) obtained with CDS and DT servers. In the docking between bioactive compounds and the RdRp protein, CDS obtained binding energy (between -8.10 and -10.60 kcal/mol) higher than DT (between -6.51 and -7.54 kcal/mol). Furthermore, docking with Mpro protein showed values in a similar range in both servers.
Referring to Mpro protein: Luteolin 7-glucoside (CDS: -8.30 kcal/mol; DT: -9.040 kcal/mol) and Syringetin glucopyranoside (CDS: -8.20 kcal/mol; DT: -8.191 kcal/mol) belonging to Turnera diffusa plant, as well as Cypellocarpin B (CDS: -9.00 kcal/mol; DT: -8.100 kcal/mol) and Cypellocarpin C (CDS: -8.60 kcal/mol; DT: -7.056 kcal/mol) from Eucalyptus camaldulensis, showed higher binding energy than Remdesivir (CDS: -8.30 kcal mol; SD: -7.919 kcal/mol). For RdRp: Luteolin 7-glucoside (CDS: -10.00 kcal/mol; DT: -6.511 kcal/mol), Syringetin glucopyranoside (CDS: -10.60 kcal/mol; DT: -7.274 kcal/mol), Cypellocarpin B (CDS: -10.10 kcal/mol; DT: -7,296 kcal/mol) and Cypellocarpin C (CDS: -9.80 kcal/mol; DT: -7.478 kcal/mol) obtained a binding energy higher than Remdesivir (CDS: -9.20 kcal/mol; DT: -7,330 kcal/mol). Since the mentioned compounds presented better binding energy obtained by Remdesivir and the other compounds, a detailed analysis of these molecules was carried out.
Cypellocarpin B and C
Cypellocarpin B (Mpro: -9.00 kcal/mol; RdRp: -10.10 kcal/mol) and Cypellocarpin C (Mpro: -8.60 kcal/mol; RdRp: -9.80 kcal/mol) with CDS showed higher binding energy to Remdesivir (Mpro: -8.30 kcal/mol; RdRp: -9.20 kcal/mol) for Mpro and RdRp. While for DT, only Cypellocarpin B (-8.100 kcal/mol) had higher binding energy than Remdesivir (-7.919 kcal/mol) with Mpro protein. In docking with RdRp protein, only Cypellocarpin C (-7.478 kcal/mol) scored higher than Remdesivir (-7.330 kcal/mol).
The literature has reported that cypellocarpins have a similar effect to (-) epigallocatechin gallate by inhibiting the activation of Epstein-Barr virus antigen, the main cause of acute infectious mononucleosis (Brezáni & Karel, 2013). In vitro studies have shown that Cypellocarpin B and C suppress cancerogenesis in mouse skin cells (Goodger & Woodrow, 2013). Furthermore, Cypellocarpin C is a potent antitumor agent with a greater effect than acyclovir when treating the HSV-2 virus responsible for genital herpes (Treml et al., 2020).
In silico studies have been carried out in which bioactive compounds of the Eucalyptus genus against SARS-CoV-2. Although, the cypellocarpins B and C molecules have not been evaluated. Fitriani et al., (2020) reported the binding energy between the Mpro protein and Cypellocarpin A with AutoDock tool 4. The docking result showed that Cypellocarpin A (-6.60 kcal/mol) has lower binding energy than Remdesivir B and C (-7.63 kcal/mol). The differences can be attributed to the number of hydrogens each molecule can donate (cypellocarpin C: 5, cypellocarpin B: 6, and cypellocarpin A: 7). The protein used for docking in the Fitriani et al. (2020) work was different. The Mpro used was 3CLpro-X77 (PDB: 6W63), and Mpro (PDB: 6LU7) was used in our work. The preparation of ligands and proteins in Fitriani et al. (2020) work was carried out with the Chimera 1.13.1 program. Hydrogens were added to the molecule, and grid box parameters were set using AutoDock Tools (ADT).
Table 1 Results of molecular coupling between bioactive molecules with CDS and DT.
| Source | PubChem CID | Molecule | Molecular weight (g/mol) | CDS | DT | ||
|---|---|---|---|---|---|---|---|
| Mpro Score (kcal/mol) | RdRp Score (kcal/mol) | Mpro Score (kcal/mol) | RdRp Score (kcal/mol) | ||||
| Chemical | 121304016 | Remdesivir (Control) | 602.6 | -8.30 | -9.20 | -7.919 | -7.330 |
| Parthenium argentatumA.Gray | 5280633 | Neochlorogenic acida | 354.31 | -7.40 | -9.30 | -7.253 | -7.075 |
| Turnera diffusa Willd. ex Schult. | 5280637 | Luteolin 7-glucosideb | 448.4 | -8.30 | -10.00 | -9.040 | -6.511 |
| 16109838 | Syringetin glucopyranosideb | 670.6 | -8.20 | -10.60 | -8.191 | -7.274 | |
| Larrea tridentata (Sessé & Moc. ex DC.) Coville | 4534 | Nordihydroguayretic acidb | 302.4 | -7.60 | -9.50 | -7.305 | -6.702 |
| Moringa oleífera L. | 5280863 | Kaempferolc | 286.24 | -7.80 | -9.30 | -8.209 | -6.641 |
| 5280704 | Apigenine 7 O-glucosidec | 432.4 | -8.00 | -9.60 | -8.910 | -7.128 | |
| Eucalyptus camaldulensis Dehnh. | 10506563 | Cypellocarpin Bd | 538.5 | -9.00 | -10.10 | -8.100 | -7.296 |
| 10625791 | Cypellocarpin Cd | 520.5 | -8.60 | -9.80 | -7.056 | -7.478 | |
| Taraxacum officinale (L.) Weber ex F.H.Wigg. | 72326 | Betuline | 442.7 | -7.10 | -9.10 | -8.886 | -7.542 |
| Bougainvillea glabra Choisy | 5280441 | Vitexinf | 432.4 | -7.90 | -8.10 | -7.596 | -7.427 |
aPiluzza et al., (2020), bGovea-Salas et al., (2017), cSaucedo-Pompa et al., (2018), dHakki et al., (2010), eDíaz et al., (2018), fAbarca -Vargas & Petricevich (2018).
The results of this study show that cypellocarpins B and C have better affinity against SARS-CoV2 Mpro protein than Cypellocarpin A and Remdesivir. These molecules can be considered for in vitro or in vivo studies.
Luteolin 7-glucoside
The binding energies of Luteolin 7-glucoside with Mpro, in CDS and DT were -8.30 kcal/mol and -9.040 kcal/mol, respectively. Remdesivir’s results were -8.30 kcal/mol and -7.919 kcal/mol, respectively. In CDS, Luteolin 7-glucoside score is similar to control. With DT, results superior to control are shown, which indicates better binding energy with Mpro protein in this server.
In RdRp, CDS binding energy was -10.00 kcal/mol; in DT was -6.511 kcal/mol for Luteolin 7-glucoside. In the case of Remdesivir, the values obtained were -9.20 kcal/mol in CDS and -7.330 kcal/mol in DT (Table 1). In CDS, Luteolin 7-glucoside score is higher than the control. In DT, the Luteolin 7-glucoside score is lower than the control.
Luteolin and its derivatives have antioxidant, antimicrobial, anti-inflammatory, and anticarcinogenic activities (Žemlička et al., 2014). Luteolin showed potent antiviral activity against SARS-CoV (Yi et al., 2004), Japanese encephalitis virus (Fan et al., 2016), HIV-1 protease (Mehla et al., 2011), Epstein-Barr virus, Rhesus rotavirus, and Chikungunya virus (Zakaryan et al., 2017).
Luteolin has an excellent binding affinity to amino acid residues of the active site of the spike protein of SARS-CoV-2 (Sen et al., 2020). A molecular docking study showed that Luteolin-7-glucoside could potentially inhibit Mpro in SARS-CoV-2 (Khaerunnisa et al., 2020), reporting a value of -8.17 kcal/mol, using Autodock 4.2; this value is close to obtained in the present study with CDS for Mpro (-8.30 kcal/mol).
Another computational study reported Luteolin-7-glucoside has optimal binding energy for Mpro inhibition (-10.66 kcal/mol with Autodock4, -8.4 kcal/mol with Autodock Vina, and -9.73 kcal/mol with Smine) (Giguet-Valard et al., 2020). These values are close to those obtained with CDS (-8.30 kcal/mol) and DT (-9.040 kcal/mol); this molecule has the potential as an inhibitor of SARS-CoV-2. However, in vitro, or in vivo studies are necessary.
Syringetin glucopyranoside
Syringetin glucopyranoside with Mpro in CDS and DT showed docking scores of -8.20 kcal/mol and -8.191 kcal/mol, respectively; for Remdesivir were -8.30 kcal/mol and -7.919 kcal/mol. In CDS, the score is lower than Remdesivir. However, this value is not far from control. In the case of DT, the results are higher than Remdesivir, indicating better binding energy with Mpro protein in this server. RdRp shows a similar trend to Luteolin 7-glucoside, with higher CDS values than Remdesivir. While with DT, lower values were reported (Table 1).
The information on Syringetin glucopyranoside is minimal; Syringetin is a flavonoid, specifically a flavonol. A wide variety of biological activities have been described in flavonoids (Brodowska, 2017; Zakaryan et al., 2017). Antiviral activity has also been reported against certain RNA viruses, such as a respiratory syncytial virus (RSV), poliovirus, and DNA viruses, such as herpes simplex virus (HSV-1) (Naithani et al., 2010). Damiana (Turnera diffusa) compounds such as Luteolin and Syringetin glucopyranoside possess anti-inflammatory, antibacterial, antioxidant, and antiviral activity (Govea-Salas et al., 2017).
In vitro antiviral activity of some flavonoids (including syringetin) against the respiratory syncytial virus (RSV) has been evaluated. The results showed the antiviral activity of flavonoids (Xu et al., 2020). Therefore, flavonoids and flavanols as antiviral agents are promising. This work is the first to investigate this molecule, so we recommend conducting a more extensive study.
The properties of these bioactive compounds influence the outcomes of viral infections (antioxidant effects that can help reduce oxidative stress levels). Their anti-inflammatory and immunomodulatory effects can greatly reduce the damage caused by viral infection. Polyphenols have inhibitory action against SARS-CoV-2 and SARS-CoV, and MERS-CoV. Interactions of polyphenols with viral proteins and host cell receptors can interfere with virus entry and replication. In vitro studies have shown that polyphenols can interrupt the viral cycle by binding to their proteins. These interactions could inhibit proteins, such as Mpro, and RdRp, or alter the binding of structural proteins, such as protein S. Curcumin is one of the molecules most studied in silico and in vitro as a potent inhibitor of SARS-CoV Mpro. Curcumin concentrations greater than 30 µg/mL can reduce the activity of SARS-CoV 2 Mpro by more than 50%, while the highest concentration of 75 µg/mL produced a residual activity of 28.1% (Gligorijevic et al., 2021). Flavonoids have also been shown to have the potential to inhibit the activity of SARS-CoV-2 viral proteins (Benarba & Pandiella, 2020).
The intake of polyphenols for the general population is 0.9 g per day; after ingestion, only 5-10% of the total polyphenol is absorbed in the small intestine, while the remaining 90-95% can accumulate in the lumen of the large intestine up to the millimolar range (Gligorijevic et al., 2021).
Orally administered polyphenols should have beneficial effects in preventing and treating COVID-19, at least in the gastrointestinal tract.After ingestion, polyphenols interact with proteins in the oral cavity. Therefore, polyphenols could inhibit SARS-CoV-2 entry and replication, reducing the risk of SARS-CoV-2 infection. In addition, a high expression of the ACE2 receptor of SARS-CoV-2 was found in the epithelial cells of the oral mucosa and the tongue, so the oral cavity is considered a high potential risk for SARS-CoV-2. (Gligorijevic et al., 2021).
Future studies on the beneficial effects of bioactive molecules such as polyphenols, tannins, and flavonoids on COVID-19 should also consider the bioavailability of polyphenols, their metabolites, and their effective concentrations to induce an effect.
Protein-ligand complex interactions
The Mpro protein consists of a homodimer with each polypeptide composed of three domains: I (residues 8-101), II (residues 102-184), and III (residues 201-303). The binding site is in a cleft between domains I and II. Its reaction center is Cys145-His164, following a mechanism similar to other coronaviruses (Pavlova et al., 2021).
Nsp12 consists of an RdRp domain (residues Ser367 to Phe920) and a nidovirus-specific N-terminal extension domain (residues Asp60 to Arg249). It folds into three subdomains namely thumb, palm, and fingers. Furthermore, Nsp12 from SARS-CoV-2 possesses a newly identified β-hairpin domain at its N-terminus (Guedes et al., 2021).
Remdesivir and RdRp binding is stabilized at the active site with four types of bonds, three hydrogen bonds with Arg475, Tyr539, and Asn611, a C-H (carbon-hydrogen) bond with Asp543, a pi cation bond with Lys471 and alkyl bond with Ala608 (Fig. 2a). Remdesivir interacts with active site residues of Mpro via H (hydrogen) bonds with Ser1 and Thr280, C-H bonds with Gly2 and Leu282 residues, a Pi alkyl bond with Phe291 and Van der Waals interactions with nine residues (Fig. 1a.) A Pi cation double bond to Lys471 residue, an alkyl bond with Ala608, a C-H bond with Asp543, and H bonds with Arg475, Tyr539, and Asn611 residues (Fig. 1a).
Furthermore, Luteolin 7-glucoside exhibits H bonds to Ser1, Phe3, Lys5, Arg4, and Asn214 residues when interacting at the active site of Mpro (Fig. 1b). In RdRp, Luteolin 7-glucoside binds via H bonds to residues Arg473, Arg475, Asp543, Ser602, Ser679, Asp680, and a C-H double bond to residue Asp543 (Fig. 2b).
Syringetin glucopyranoside interacts with Mpro and RdRp at its active site with only H and C-H bonds. The interactions with Mpro are H bonds with Lys5 and Asp216 residues, C-H bonds with Leu282 and Gly283, and Phe3 residues forming both types of bonds (Fig. 1c). Docking with RdRp shows C-H bond interactions with residues Asp543 and Asp680, and H bonds with Ile468, Arg473, Arg475 (double bond), Cys542, Asp543, Asp681, and Ser734 (Fig. 2c).
Cypellocarpin B binds to the active site of Mpro protein via H bonds with Arg4, Phe3, and Asp216, Van der Waals interactions with Gly2 Asn214, Leu282, Thr280 residues, and C-H bonds with Gly283 (Fig. 1d). Cypellocarpin B with RdRp via four conventional hydrogen bonds to residues Lys471, Arg475, Asp680 and Ser734, Van der Waals forces at residues Arg473, Asn611, Ala608, Leu678, Tyr539, Trp537, Asp538, and cationic C-H and Pi bonds to residues Ser679 and Asp681, respectively (Fig. 2d).
In Cypellocarpin C, the interactions with Mpro protein were formed by Asp216 residues through H bonds, the Gly283 residue through C-H bonds, and Arg4, Thr280, and Leu282 forming Van der Waals interactions (Fig. 1e). Cypellocarpin C is stabilized at the active site of RdRp protein via H bonds (Ser469, Arg475, Cys 542, Asp543), Van der Waals interactions (Arg756, Ala467, Ile 468, Ala470, His359, Ser734, Arg473, Lys541, Arg 544, Asp680), and a double bond (Pi cation and Pi alkyl) in Lys471 residue (Fig. 2e).
Although most interactions show variations in bond types and residues, RdRp, when interacting with Remdesivir, Luteolin 7-glucoside, Syringetin glucopyranoside, and Cypellocarpin B and C, shows a typical hydrogen bond type interaction with residue Arg475.
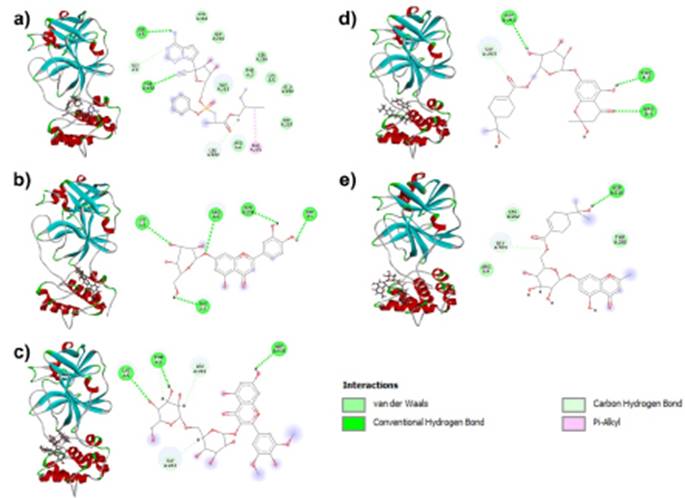
Source: Own elaboration based on Discovery Studio Visualizer. Remdesivir, b) Luteolin 7-glucoside, c) Syringetin glucopyranoside, d) Cypellocarpin B, e) Cypellocarpin C. The interactions of the molecules with the amino acid residues of the proteins are observed.
Figure 1 Three-dimensional structure and 2D interactions of SARS-CoV-2 Mpro.
Analysis of docking servers
Seven molecules analyzed for Mpro by CDS (Remdesivir, Neochlorogenic acid, Syringetin glucopyranoside, Nordihydroguayretic acid, Cypellocarpin B, Cypellocarpin C, and Vitexin) showed a higher energy value, compared to DT. When performing the docking between the molecules and Mpro, DT showed that five of the bioactive compounds had higher binding energy (-9.040, -8.191, -8.209, -8.100, -8.910, and -8.886 kcal/mol) than Remdesivir (-7.919 kcal/mol), while on CDS server, only two compounds showed higher binding energy (-9.00 and -8.60 kcal/mol) than the control (-8.30 kcal/mol). Regarding RdRp, all the scores obtained by CDS were higher than DT.
In the docking with RdRp, the CDS server results showed that eight of the molecules had higher scores (-9.30, -10.00, -10.60, -9.50, -9.30, -10.10, -9.80 and -9.60 kcal/mol) than Remdesivir (-9.20 kcal/mol). In contrast, the DT server presented only three scores (-7.478, -7.542, and -7.427 kcal/mol) higher than Remdesivir (-7.330 kcal/mol). Differences in binding energy values between servers may be mainly due to the different algorithms or docking engines used on each server and differences in default viral protein structures in each server.
For small molecule docking, CDS uses Autodock Vina as a docking engine (Kong et al., 2020). Excellent agreement has been shown between docking scores obtained from CDS and manual docking using AutoDock Vina (Sen Gupta et al., 2020), making this server a valuable tool for screening molecules of interest against SARS-CoV-2. However, in vitro or in vivo confirmatory studies are recommended.
The DT server uses the DT program as the docking engine. The web server uses the computing facilities of the Brazilian high-performance platform (SINAPAD) and the SDumont supercomputer (da Silveira et al., 2019; Guedes et al., 2021). DT has been evaluated with other molecular docking programs for the efficiency of scoring functions (Rerank > MolDock> PLANTS> AutoDock Vina> DT) (González-Paz et al., 2020), concluding that DT is inferior in this aspect to AutoDock Vina (the engine used by CDS).
In addition, there are differences when performing the analysis; for example, the box size in CDS is 30 x 30 x 30, and in DT, it is 20 x 20 x 20. Also, in CDS 10 runs are made, and thetop 11 best run is selected; in DT 24 runs are carried out, and the final value is taken.
Some studies have obtained promising results for DT compared to other docking programs such as Glide, GOLD, and AutoDock Vina (considering various molecular targets and chemical classes of ligands). DT is a promising server for testing molecules against SARS-CoV-2; however, in vitro or in vivo confirmatory testing is recommended as with CDS. This server has great potential to be widely used in receptor-ligand studies (Santos et al., 2020).
Referring to each server’s proteins, the default Mpro protein in DT is a dimer of two homologous amino acid chains (A and B). CDS uses a monomeric structure to do the docking. Both servers have used the structure reported by Jin et al. (2020) (PDB 6LU7). With RdRp, both servers use the RdRp structure reported by Yin et al. (2020) (PDB 7BV2).
A complete analysis of the CDS and DT servers is required, as well as the proteins each have to perform molecular docking. However, current tools can be considered for rapidly screening molecules with potential activity against SARS-CoV-2.
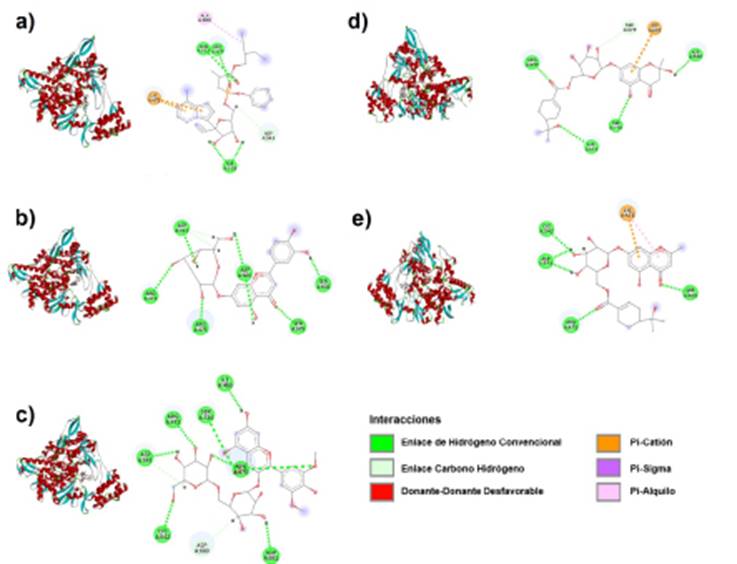
Source: Own elaboration based on Discovery Studio Visualizer. a) Remdesivir, b) Luteolin 7-glucoside, c) Syringetin glucopyranoside, d) Cypellocarpin B, e) Cypellocarpin C. The interactions of the molecules with the amino acid residues of the proteins are observed.
Figure 2 Three-dimensional structure and 2D interactions of the RdRp protein of SARS-CoV-2.
Study limitations
In silico studies indicate that there are molecular interactions with the protein of interest. However, this method cannot determine data such as the concentration that generates the interaction: in vitro and in vivo experiments are recommended to verify biological activities. Furthermore, the efficacy and safety must be further studied in vivo and validated in COVID-19 patients. The bioavailability, modes of administration, safe doses, exposure time, and pharmacokinetic profile of the molecules must also be considered.
Although in silico approaches do not guarantee antiviral behavior. These are the first steps for future in vitro and in vivo studies of molecules containing antiviral activity against SARS-CoV-2 and other kinds of virus.
Study limitations
In silico analysis indicates the interaction of a molecule or binding energy with the protein of interest. However, data such as the concentration that induces the interaction by this method remains unknown and must be complemented by in vitro and in vivo experiments to verify biological activities.
Possible genomic variations in the binding site region of molecular targets can drastically affect the binding mode and affinity of ligands and alter the identification of promising compounds. The clinical utility of these molecules remains to be demonstrated, as current data are premature. In addition, efficacy and safety should be further investigated in vivo and validated in patients with COVID-19, taking into account the bioavailability, modes of administration, safe doses, exposure time, and pharmacokinetic profile of the molecules.
While in silico approaches do not necessarily guarantee antiviral behavior, they are the first step for future in vitro and in vivo studies of molecules with antiviral activity against SARS-CoV-2 and other types of viruses.
Conclusions
The results revealed that cypellocarpin B, cypellocarpin C, luteolin 7-glucoside, and syringetin glucopyranoside have the best affinities to SARS-CoV-2 proteins, Mpro and RdRp compared to remdesivir. The molecular docking results confirm that the molecules present in medicinal plants from Mexico are options for treatment against COVID-19. However, in vitro or in vivo studies are recommended to demonstrate the safety and efficacy of these compounds. CDS and DT are useful tools for evaluating molecules with potential activity against SARS-CoV-2.

