











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkANTECEDENTES
El carcinoma de conductos colectores es un tumor renal poco frecuente descrito en 1979.1 Según la cohorte más grande reportada por la National Cancer Database (NCDB), se estimó una incidencia de 0.2% y supervivencia media de 13.2 meses.2 Su histogénesis corresponde a células de los conductos colectores en la médula adrenal. De acuerdo con su origen embriológico, el carcinoma de los conductos colectores inicia en la yema ureteral y el carcinoma medular en el blastema metanéfrico.
El 90% de los pacientes manifiesta síntomas y enfermedad a distancia similares a los que provoca el cáncer renal, además de pérdida de peso. El diagnóstico se establece después de analizar la pieza de la nefrectomía; sin embargo, algunos autores recomiendan obtener una biopsia previa al procedimiento quirúrgico, debido al alto riesgo de mortalidad perioperatoria.3,4
CASO CLÍNICO
Paciente femenina de 77 años de edad, con antecedente de diabetes tipo 2, nefropatía secundaria (KDIGO 3A) e hipertensión arterial sistémica de larga evolución, sin complicaciones. Inició su cuadro clínico con dolor en el flanco izquierdo, de 20 días de evolución, además de dolor óseo. Después de la exploración física se solicitó un ultrasonido, que reportó una masa dependiente del polo superior del riñón izquierdo. La tomografía simple, complementada con resonancia magnética, evidenció una lesión de 5.7 x 7.3 cm, de consistencia sólida, con necrosis central y extensión hacia el seno renal (T2a; GII) (Figura 1). Se descartó metástasis a hueso por gamagrafía ósea. Los estudios de laboratorio de ingreso documentaron anemia microcítica hipocrómica.
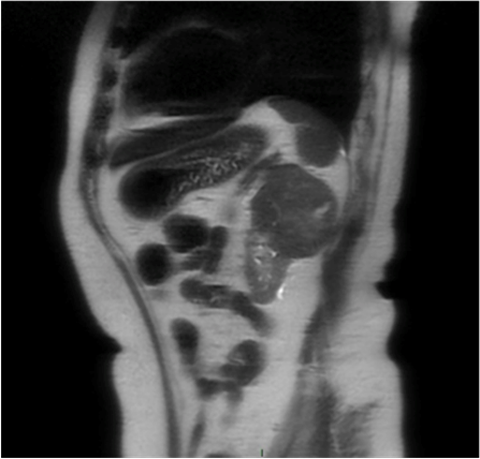
Figura 1 Resonancia magnética. Corte sagital que muestra una lesión sólida en el riñón izquierdo, con necrosis central.
El tratamiento consistió en nefrectomía radical laparoscópica izquierda, cuya pieza histopatológica reportó un tumor de 9 cm, que afectaba el polo superior y medio del riñón izquierdo. La neoplasia mostró superficies blancas, sólidas y multinodulares sin invasión del seno renal; microscópicamente se observó una neoplasia epitelial maligna, formada por túbulos de luces irregulares bordeadas por células con marcado pleomorfismo celular y nuclear (células en estoperol) con márgenes negativos. Los resultados fueron sugerentes de carcinoma de conductos colectores de Bellini (Figura 2). Durante el seguimiento, a 5 meses, la tomografía computada simple mostró un nuevo ganglio latero-aórtico izquierdo de 16 mm, cercano al lecho quirúrgico. Por sospecha de recurrencia se realizó escisión de la lesión, además de linfadenectomía retroperitoneal abierta izquierda que reportó la coexistencia de 20 ganglios con hiperplasia linfoide negativa a malignidad. La paciente se mantuvo estable y en vigilancia. La tomografía computada documentó probable linfocele en el lecho quirúrgico, que se manejó con vigilancia estrecha. Treinta y nueve meses después de la nefrectomía, la tomografía computada evidenció dos tumores retroperitoneales: uno paraaórtico y otro intercavoaórtico, con ejes mayores de 4 y 5 cm, respectivamente, compatibles con recurrencia de la enfermedad (Figura 3).
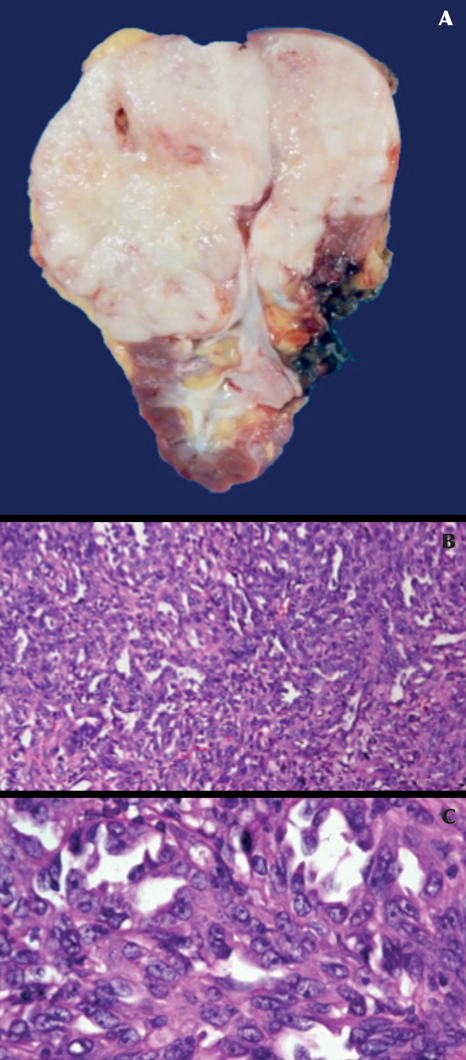
Figura 2 Tumor de 9 cm en el polo superior y medio del riñón izquierdo con superficies blancas, sólidas y multinodulares sin afección del seno renal (A). Microscopia que muestra túbulos de luces irregulares bordeadas por células con marcado pleomorfismo celular y nuclear (células en forma de “estoperol”) (B y C).
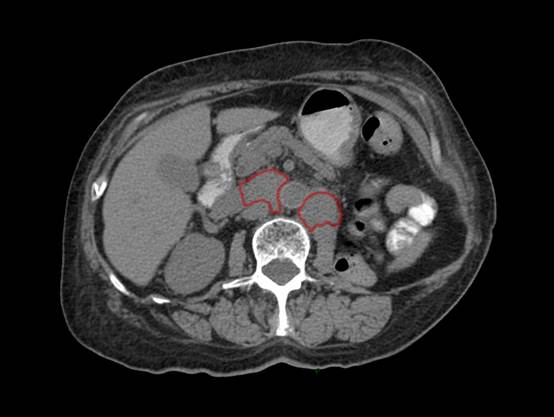
Figura 3 Tomografía computada. Corte axial que evidencia dos lesiones (rojo) en el espacio intercavoaórtico y paraaórtico, de 7 y 5 cm, respectivamente.
Se llevó a cabo metastasectomía, que histopatológicamente reportó un tumor sólido, irregular, de color blanco-grisáceo, con áreas extensas de hemorragia en su porción inferior izquierda y focos discretos de necrosis (Figura 4).
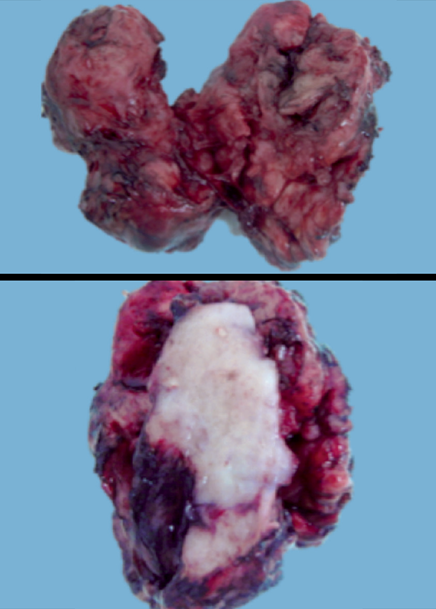
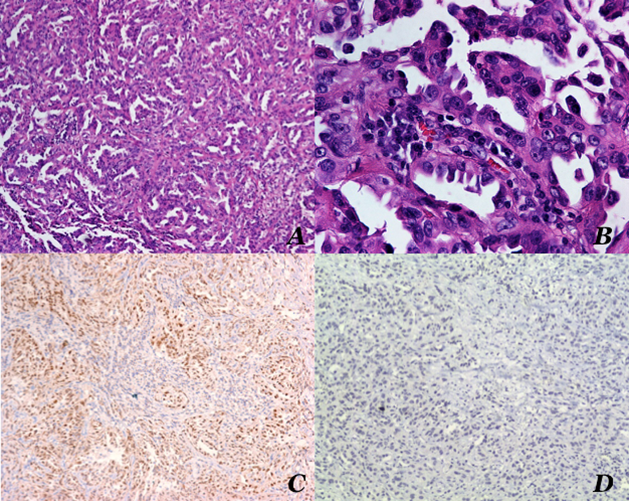
Hoy día, después de 42 meses de seguimiento, la paciente se mantiene en vigilancia y sin datos de recurrencia. Microscópicamente, con los objetivos de bajo aumento, se observó un patrón de crecimiento con cordones entrecruzados, con espacios vacíos angulados, en forma de hendiduras o rendijas; con los objetivos de mayor aumento las células neoplásicas mostraron pérdida de la polaridad, algunas de ellas parecidas a la forma de un “estoperol”, compatible con recurrencia de carcinoma renal de conductos colectores (Figura 5). Los núcleos se observaron grandes y ovales; cromatina abierta y rechazada a la periferia y nucléolo prominente. La inmunorreacción resultó positiva para PAX8 y negativa para p63. La paciente tuvo evolución clínica favorable y egresó estable.
DISCUSIÓN
El carcinoma de conductos colectores es una de las neoplasias renales más raras, cuyas manifestaciones y diagnóstico suelen establecerse en estadios avanzados de la enfermedad. La función de la quimioterapia, radioterapia e inmunoterapia han demostrado beneficios limitados y sujetos a discusión.5 Sólo un estudio retrospectivo ha reportado disminución de la mortalidad cuando se complementa con cirugía en pacientes con metástasis.2
Puesto que se trata de un tumor excepcional y el diagnóstico se establece en etapas avanzadas, le confiere un mal pronóstico. Un estudio de 41 casos pareados con carcinoma de células claras reportó una tasa de supervivencia similar al de pacientes con carcinoma de conductos colectores de Bellini.6
Los sitios de metástasis más frecuentes son los nodos, las glándulas adrenales, el hueso, pulmón e hígado.1 El pronóstico de supervivencia varía de 11 a 15 meses; sin embargo, dos estudios señalan un caso de 58 (pT1) y otro de 120 meses (T2) con diagnóstico incidental de la enfermedad. De acuerdo con la bibliografía revisada, el caso aquí reportado corresponde al de mayor supervivencia y tiempo de recurrencia descrito en un caso de T2M0 sintomático: 39 meses.7
La media de recurrencia local es de 4.9 meses (límite máximo de 10 meses).3,8 En la paciente de este estudio se reportó una recurrencia local de 39 meses, con adecuado control oncológico mediante resección quirúrgica.

