











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkThe domestication process and its effect on genomes. During the Neolithic, a major change occurred in the way humans obtained food and resources. Human transitioned from hunters-gathers to practice agriculture, a sedentary lifestyle, settlement of villages and the creation of ceramic, eventually leaded to the development of hierarchical civilizations (Gepts 2014, Larson et al. 2014). During this process, domestication was a key technological tool (Gepts 2014).
Domestication is an evolutionary process, in which humans promote the adaptation of wild species to agroecological niches and to local preferences (Figure 1. Casas et al. 1996, Larson et al. 2014). The evolutionary trajectory from wild to domesticated population is a complex multi-stage process, that involves several steps (Figure 2. Meyer & Purugganan 2013):
Predomestication: humans begin to purposely plant and look after wild plants with favorable traits.
Artificial selection in environments created by humans: at this the stage, human selective pressures promote alleles related with domestication traits.
Spread and diversification: propagation and local adaptation to different agroecological and cultural environments. In this stage, cultivated and wild populations diverge and domesticated traits diversify.
Deliberate breeding: this last stage has been practiced intensively in the last century. In classical breeding, controlled crosses between inbred lines are made and then individuals with desirable traits are selected. In the last decades, molecular and genetic tools have been integrated to breeding practices.

Figure 1 Examples of wild and domesticated forms of crops. The first image of each row is the wild relative. a) teosinte and maize (Zea mays); b) chilli pepper (Capsicum annuum); c) common bean (Phaseolus vulgaris); d) cotton (Gossypium hirsutum). Images taken from CONABIO.and CIAT and CIAT.
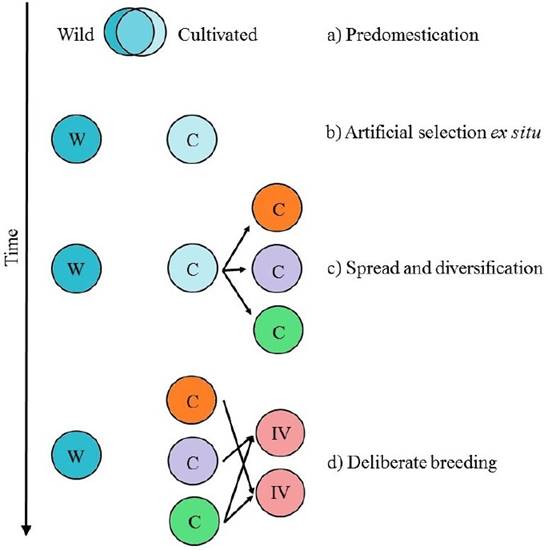
Figure 2 Evolutionary stages of domestication. W represents wild populations, C cultivated populations and IM improved lines. a) Predomestication; b) Management in environments created and controlled by humans; c) Spread and adaptation of cultivated populations; d) Deliberate breeding. It is important to notice that these stages are not mutually exclusive and can occur in presence of wild progenitors, increasing the probability of gene flow. Modified from Meyer & Purugganan (2013).
These stages are not mutually exclusive and can occur simultaneously, even today. For example, Nahuas communities in Mexico consume wild progenitors of crops like common bean, chili pepper, agave and tomato (in total 68 wild species) using techniques and tools that are available since the Archaic period (Zizumbo-Villarreal et al. 2012).
The domestication process affects the genomes of cultivated species and leaves a signature that can be identified. Domestication results in a pronounced alteration in the diversity and differentiation of few genes, while most of the genome reduces its variation as a result of population bottlenecks that occur in the initial predomestication stage (Charlesworth et al. 1997, Gepts 2004).
What do domesticated plants have in common? While the domestication process has varied in different plant groups depending on the mating system, growth form, harvested plant parts and economic importance there are several aspects that have been identified to be common to most domestication processes. These include loss of genetic diversity, the genes associated with the domestication process, an accumulation of deleterious variants, the relation between the cultivar and its wild relatives and the possibility of phenotype convergence and its molecular basis in different domestication processes.
Genetic diversity reduction.- One of the most important determinants in crop evolution is the level of genetic diversity contained in the domesticated accessions, especially with reference to the wild ancestral gene pool. Genetic diversity is important as a necessary condition for further evolution in response to selection pressures, not only in the wild but also in breeding programmes (Gepts 2002). Genetic diversity reduction has been widely described in some of the most important crops. It is due to genetic drift resulted from population bottlenecks, and to artificial selection and the consequent selective sweep (Charlesworth et al. 1997, Gepts & Papa 2003, Gepts 2014). The domestication process itself, the spread from the center of origin and modern breeding practices, involve population bottlenecks because only some genotypes are selected, promoting a genetic diversity reduction (Gepts & Papa 2003). This pattern has been reported in numerous species, for example, Mesoamerican common bean (~20 % reduction; Schmutz et al. 2014); sunflower (~30 %; Renaut & Rieseberg 2015); soybean (~30 %; Li et al. 2013); maize (~17 %; Hufford et al. 2012); and in cultivated Agave species (from 21 to 66 %; Eguiarte et al. 2013). In sunflower, maize and soybean, the largest diversity decrease occurs between wild populations and landraces; on the contrary, the difference in genetic variation between landraces and improved varieties is low, indicating that breeding practices, at least in these cases, do not have major effects in terms of genetic diversity, at least in these species (Li et al. 2013, Hufford et al. 2012, Renaut & Rieseberg 2015).
However, exceptions to this pattern have been reported. For example, in carrots, high levels of gene flow between wild and domesticated populations, besides a strong inbreeding depression, have prevented from genetic variation reduction (Iorizzo et al. 2013). In the Andean region, cultivated common bean is more genetically diverse than wild populations (Schmutz et al. 2014). In central Mexico, managed populations of the cacti Stenocereus stellatus (Pfeiff.) Riccob., presents higher levels of genetic variation than unmanaged populations, showing that the management made by humans can potentially alter the genetic diversity of non-domesticated species (Cruse-Sanders et al. 2013).
Mutations in genes under domestication-. Identification and isolation of genes that underlie domestication traits provide the opportunity to identify patterns present in these loci. These genes show a wide range of functions, from transcription factors to metabolic enzymes, although many encode similar enzymes or are involved in the same metabolic pathways (Meyer & Purugganan 2013).
According to Meyer & Purugganan (2013), the most frequent functional changes occur in alleles involved in function loss and gene expression. These mutations promote major effects in phenotypes, which usually distinguish domesticated and wild populations. For example, in woodland strawberry (Fragaria vesca L.), a 2-bp deletion in the coding region of the KSN gene –which is a transcriptional factor– introduces a frameshift, resulting in continuous flowering (Iwata et al. 2012). Also, the function loss of Vrs1 gene in barley (Hordeum vulgare L.), changed the inflorescence architecture from two-rowed to six-rowed type (Komatsuda et al. 2007).
Mutations in genes under domestication include single nucleotide polymorphisms (SNPs), insertions and deletions (indels), transposon insertions, gene duplication (including gene copy number) and chromosomal rearrangements. The most frequent alterations are SNPs, followed by indels (Meyer & Purugganan 2013).
An important question is whether mutations that lead to domestication phenotypes are new or preexisted in wild populations. This has implications in the nature of selective sweeps and crop species evolution dynamics; for example, a selective sweep caused by a preexisting mutation leaves weaker signatures of selection and allows rapid evolution, because it has a higher frequency than a new mutation (Gepts 2014).
There are some examples of alleles that underlie domesticated traits that appeared recently and are not present in wild populations. That is the case of LG1 gene in rice, associated with closed panicle (Huang et al. 2012) or the SUN gene duplication in tomato, which regulates fruit shape (Rodriguez et al. 2011). However, there are cases of alleles that preexisted in wild populations at low frequency that were subjected to human selective pressures. For example, the Brassica oleracea CAL gene encodes a transcription factor that regulates floral meristem development, and a nonsense mutation leads to the proliferation of floral meristem in domesticated cauliflower and broccoli (Purugganan et al. 2000). This mutation is either fixed or present at a high frequency in cultivars, but it is also present at low frequencies in wild populations (Purugganan et al. 2000). Another example is the single-stem phenotype of domesticated maize, which is controlled by the dominant Tb-1 allele. This allele arose before domestication by the insertion of a Hopscotch transposon leading to overexpression. The insertion occurred 28,000-23,000 years BP, predating domestication (Studer et al. 2011). This suggests that many domesticated traits arise not from new mutations but rather from mutations that segregate in wild population of crop species.
Accumulation of deleterious mutations.- Mutations can have several effects on fitness that range from lethal to neutral and advantageous, but most of these new variants in coding regions are expected to be deleterious, because they may alter phylogenetically conserved sites or can cause loss of protein function (Kono et al. 2016). Under mutation-selection balance and in sexually reproducing species, the accumulation of deleterious alleles -- particularly strongly deleterious and with dominant effects -- is infrequent, because recombination brings together deleterious variants, resulting in unfit genotypes that are eliminated from the population, purging deleterious mutations by purifying selection. Weakly deleterious variants on the other hand, can be potentially maintained under some circumstances, like population size reduction and/or inbreeding, reducing the effective recombination rate and promoting the accumulation of deleterious mutations (Morrell et al. 2011, Renaut & Rieseberg 2015).
Besides, as a species expands into new environments (either natural or artificial), genetic drift could increase, due to the reduction of effective population size and a posterior fast growth rate. During the process of domestication and improvement, populations undergo multiple bottlenecks, accompanied by strong artificial selection and the relaxation of selective pressures on traits important in the wild (Morrell et al. 2011). These bottlenecks increase the probability that a deleterious variant increases its frequency and gets fixed (Mezmouk & Ross-Ibarra 2014). Also, the linkage between desirable beneficial and deleterious mutations may reduce the efficiency of selection to eliminate the latter, contributing cumulatively to fitness reduction and probably constraining crop yield (Renaut & Rieseberg 2015).
The selective and demographic processes of domestication have led to three hypotheses about the patterns of deleterious mutations (Kono et al. 2016):
There will be more deleterious alleles in domesticated than in wild relatives.
Deleterious variants will be enriched near loci that have been under artificial selection.
There will be less deleterious mutations in elite cultivars than in landraces due to strong selection for yield.
There is evidence that supports these hypotheses, as it is estimated that 20 % of non-synonymous variants in rice (Lu et al. 2006) and in maize (Mezmouk & Ross-Ibarra 2014) present deleterious effects. In sunflower, landraces and elite lines possess more non-synonymous SNPs compared to wild relatives, particularly regions with low recombination rates and less deleterious mutation, are present in elite cultivars compared to landraces (Renaut & Rieseberg 2015). Also, hundreds of deleterious mutations have been identified in barley and soybean cultivars, being non-sense mutations the least frequents (Kono et al. 2016).
Recent advances in sequencing technologies permit to use bioinformatics tools to examine the patterns of deleterious variants in populations. Pure bioinformatic approaches use sequence conservation to infer variants with a significant probability of being deleterious. As a consequence, the identification and elimination of these alleles can potentially provide a complementary approach to breeding practices if identified variants are truly deleterious (Kono et al. 2016).
Gene flow between domesticated plants and their wild relatives.- Gene flow between crops and their wild relatives is common, since these two types of population coexist in sympatry, and in most cases, crops and their wild progenitors belong to the same biological species (Gepts 2014). The potential consequences of gene flow are diverse. For example, gene flow between wild and domesticated populations can result in diversity recovery in cultivated forms, as has been described in maize due to continuous introgression with teosinte (Hufford et al. 2012). This is also the case in grapevine during cultivars spread through Europe (Myles et al. 2011). Also, emergence of weeds has been reported, as occurred in hybrid rice populations from China (Jiang et al. 2011, Xia et al. 2011) and the United States (Olsen et al. 2007).
Despite gene flow, wild and domesticated plants remain phenotypically distinct, at least with respect to domesticated syndrome traits, probably because of human selective pressures on cultivated populations, and natural selection of wild forms. For example, gene flow among wild and cultivated squash taxa have been detected in Mexico (Montes-Hernández & Eguiarte 2002). But farmers do not select seeds from the individuals that present intermediate morphological characters for seed stock (Montes-Hernández et al. 2005).
Following the definition in its broad sense, introgression is the transfer of genes between genetically distinguishable populations (Rieseberg & Carney 1998). Introgression can take place between populations of wild species (wild–wild) that are related and between a cultivated species and its close wild relatives (crop–wild; Rieseberg & Carney 1998). Nevertheless, introgression is not uniform across the genome and it is unlikely to occur on regions close to genes related with domestication (Gepts 2014). Gene flow may also be asymmetric, although the direction is not consistent: for example, in common bean it mainly occurs from domesticated to wild types (Papa & Gepts 2003), but the opposite happens in maize (from Zea mays L. subspecies mexicana teosinte to cultivated maize; Hufford et al. 2013).
Introgression may produce profound effects on domestication evolutionary trajectories. The evolutionary history of rice (Oryza sativa L.) is an example: Molina et al. (2011) suggest that rice domestication occurred in China, where japonica variety arose. Later, indica variety was originated by a hybridization event between japonica and a putative indica protoform or with O. ryfipogon from South Asia. This resulted in the introgression of japonica domesticated genes.
Another case that illustrates the impact of gene flow occurs in domesticated citrus species. Wu et al. (2014) sequenced and compared the genomes of some Citrus species and showed that cultivated types derive from two progenitor species: introgression from C. maxima (Burm.) Merrill to ancestral mandarin species C. reticulata Blanco originated tangerine; sweet orange is the offspring of previously admixed individuals of these two species; and sour orange is an F1 hybrid of pure C. maxima and C. reticulata parents. The exception is pomelo, which is the result of selection on C. maxima (Wu et al. 2014).
Phenotypic convergence.- The Law of Homologous Series, formulated by Vavilov (1922), and ‘analogous variation’ term used by Darwin, are based on the observation that similar phenotypes were selected during domestication. This leads to the question: phenotypic parallelisms result from molecular convergences? In other words, selection on particular traits affects the same genes in different species? If this is true, parallelisms potentially can be used in breeding programs focusing on regions previously identified to be related with important domestication traits (Pickersgill 2009). However, contradictory data has been found and there is evidence for and against this. For example, Grube et al. (2000) found five clusters of resistance genes common to potato, tomato and chili pepper, and seven clusters common to two of these three crops; they concluded that the position of resistance genes, is conserved in Solanaceae. However, they found only two examples in which these homologous genes controlled resistance to the same disease. Therefore, knowing the sequence of alleles and the structure of their products can be useful in the development of new pathogens or pest control methods (Pickersgill 2009). Another example is the Shattering gene (Sh1), which avoids dehiscence in maize, rice, sorghum and common bean; furthermore, there is evidence that Sh1 independently emerged three times in sorghum (Lin et al. 2012, Schmutz et al. 2014).
On the other hand, evidence against molecular convergence in crops includes two cotton species (Gossypium hirsutum L. and G. barbadense L.), which were independently domesticated, but selection by humans acted in different genetic components that control fiber development (Hovav et al. 2008). Another case where parallelism in phenotypes does not correspond at the gene level occurs between common bean and pea: p and v genes are partially responsible of dehiscence inhibition in common bean, whereas dpo1 and dpo2 genes control this trait in pea, despite p and v genes are also present (Weeden 2007). Also, in common bean –which was independently domesticated in South America and Mesoamerica–Schmutz et al. (2014) identified 1,835 candidate genes under artificial selection in Mexican populations, and 748 in Andean ones, however only 59 were shared genes between the two gene pools. Probably, in some cases, genetic architecture behind domestication is so diverse that similar phenotypic changes are due to selection on different genes.
Using molecular markers to study domestication and advance breeding programs. Before the development of molecular markers, farmers and breeders only had phenotypic traits to select desirable individuals to interbreed. Nevertheless, relative long periods of time and several generations were required to evaluate and select useful genotypes. Some decades ago, molecular markers started to be used in breeding programs and in the development of new cultivars or varieties (Kim et al. 2015).
Following the first molecular markers tools (He et al. 2014), Sanger DNA sequencing allowed the identification of variants at the single base pair resolution. However, sequencing small genomes or large regions from complex genomes using Sanger technique is expensive and laborious. The Human Genome Project encouraged the development of techniques for sequence whole genomes at low cost and less time-consuming. These sequencing tools are known as massive parallel sequencing (MPS), also called Next Generation Sequencing (NGS) and have allowed the routine use of single nucleotide polymorphisms (SNPs) in the last decade, even in no model species (Escalante et al. 2014). These massive sequencing tools are used by breeders to sequence large populations, study genetic composition of crop varieties, understand evolutionary relationships between cultivars and wild relatives, identify genotype-genotype (G × G) and genotypes-environment (G × A) interactions, and increase the resolution of gene and quantitative trait locus (QTL) discovery, providing the basis for modeling complex genotype-phenotype relationships at the whole-genome level (Figure 3. Cobb et al. 2013, Varshney et al. 2014).

Several methods are used to identify relationships between genes or genomic regions and domestication syndrome traits. These can be classified in bottom-up and top-down approaches (Figure 4).
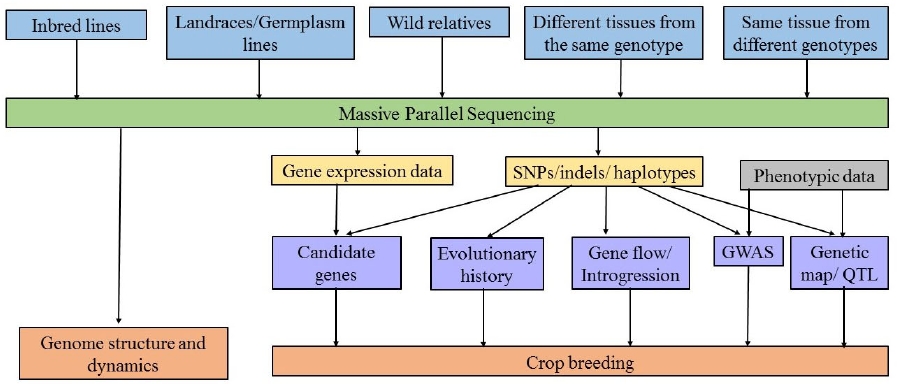
Figure 4 Approaches used to identify genes or genomic regions under selection: Top-down vs. bottom-up.
Top-down domestication studies.- Classical studies to infer the genetic basis of domestication traits use a top-down approach, especially when used in mapping QTLs. Traditional QTL mapping studies begin with two inbred parental lines that differ in their trait values. After creating an F1 generation by crossing the parental lines, F1 individuals are crossed to create an F2 mapping population. These individuals are measured for the trait of interest and genotyped at markers that segregate between parental populations. QTLs are then identified by correlations between trait value and genotype at a locus. The ability to identify a QTL using this method depends on the scale of recombination in the mapping population. For this reason, recombinant inbred lines (RILs) are often used to increase mapping resolution (Mackay et al. 2009).
On the other hand, genome-wide association studies (GWAS) utilize association mapping, also known as linkage disequilibrium (LD) mapping, to map QTLs by taking advantage of historic LD to identify statistically significant phenotype-genotype associations (Varshney et al. 2014). Unlike classical methods to identify QTLs, in GWAS population mapping is usually an outbred population and the markers used are SNPs. In general, the results are better than the ones using RILs. However, the power to detect a QTL depends of the phenotypic variance that is explained by the QTL that is determined by its effect and allele frequency. Because of this, GWAS generally has low power to detect QTL variants with small effect or that are rare in the population (Mackay et al. 2009). Nevertheless, GWAS have been successfully performed in several crops, and the use of massive sequencing makes possible to genotype larger populations with a higher density of markers (tens to hundreds of thousands of markers are needed in GWAS), increasing mapping resolution (Varshney et al. 2014, Andrews et al. 2016).
Results of the top-down approach have shown that many traits are controlled at least in part by a few major genes, that genetic effects predominate over environmental ones, and that some genes are linked. It is likely that this approach underestimates the number of genomic regions involved in domestication syndrome. For example, Hufford et al. (2012) estimated that only ~3 % of maize genome have been affected by domestication using top-down approach.
The disadvantages of the top-down approach include that phenotypic information is required, and that only few traits can be considered at the same time (Tang et al. 2010). A recent study using a top-down approach was performed in rice, in which 203 varieties with well-characterized phenotypes were sequenced, and three traits were mapped (Wang et al. 2016). For amylose content and seed length traits, this approach leads to direct identification of the previously identified causal SNPs as the major-effect loci. For pericarp color, they identified a new major-effect locus. Although previously known loci can explain color variation in the varieties of two main subspecies of Asian domesticated rice, japonica and indica, the new locus identified is unique to another domesticated rice subgroup, aus, and together with existing loci, can fully explain the major variation in pericarp color (Wang et al. 2016).
Bottom-up domestication studies.- Compared to classic genetic studies, in which relatively few markers are used, genomic approaches provide additional information about the diversity and LD patterns at the genome level. Therefore, genomics provides a broader view across taxa and across individual genomes (Gepts 2014). The bottom-up approach consists in population genetic screenings of genome-wide diversity, including sequence diversity departures from neutrality, scans for selective sweeps or highly divergent regions (FST). Further research, involving functional genetics, is then needed to directly establish a causal effect between candidate genes or alleles and phenotypes.
Comparison of sequence diversity combined with neutrality tests in wild and domesticated populations can identify regions that were affected by domestication. Furthermore, comparisons between landraces and wild forms focus specifically on the effect of initial domestication, whereas comparisons between landraces and improved cultivars measure the effect of subsequent selection, including modern breeding (Tang et al. 2010, Gepts 2014). The bottom-up approach does not require previous phenotypic data, allowing the identification of a larger amount of candidate genes or genomic regions under selective pressures, unlike the top-down approach which can only consider few genes or regions (Tang et al. 2010). For example, with a top-down approach, in which only ~3 % of the maize genome was estimated to be affected by domestication, using a bottom-up approach 1,764 genes (representing ~6.6 % of its genome) were inferred to be related to domestication and 1,508 genes (~5.6 %) related to breeding (Hufford et al. 2012).
An example of selective sweep detection using a bottom-up approach includes sorghum, where selective sweeps that encompass genes for starch synthesis enzymes, seed shattering, plant height and time to maturity, were detected using 1,000 accessions and ~265,000 SNPs (Morris et al. 2013). Another example was found in rice, where by resequencing 1,529 wild and domesticated accessions, 55 selective sweeps were detected, including Bh4 (hull color), PROG1 (tiller angle), sh4 (seed shattering), qSW5 (grain width) and OsC1 (leaf sheath and apiculus color; Huang et al. 2012).
Challenges to overcome using a bottom-up approach include that the phenotypic traits affected by the genes or regions encompassed in selective sweeps may be and remain unknown, and that demographic events may hide selective signatures because they alter diversity patterns, and standard test typically assume that populations evolve according to the idealized Wright-Fisher model, with panmictic populations of constant size (Ross-Ibarra et al. 2007). When these assumptions are inaccurate, because domestication involves genetic bottlenecks followed by demographic expansion, tests to detect selection can be inaccurate (Ross-Ibarra et al. 2007).
Advantages of using molecular markers.- There are two main types of genomic-assisted breeding: marker-assisted selection (MAS) and genomic selection (GS). MAS, which includes marker-assisted back-crossing (MABC), uses molecular markers that map within specific genes or QTLs known to be associated with target traits or phenotypes to select individuals that carry favorable alleles and/or discard those that do not (Mackay et al. 2009).
GS uses all available marker data for a population as predictor of breeding value. GS integrates marker data from a training population with phenotypic and when available, pedigree data to generate a prediction model. The model output genomic estimated breeding values (GEBVs) for all genotyped individual, which are used as a predictor of how well a plant will perform as a parent for crossing (Varshney et al. 2014).
The advantage of using molecular markers in breeding programs is that genotypic data obtained from a seed or seedling can be used to predict the phenotypic performance of mature individuals without the need for extensive phenotypic evaluation over long periods of time, allowing for more selection cycles and greater genetic gain per unit of time (Varshney et al. 2014). Previously, marker data were expensive per data point, and laborious to generate, and MAS were constrained by the number of available markers. As a result, only markers in important genome regions were utilized to predict the presence or absence of agriculturally valuable traits. By contrast, the use of massive parallel sequencing technologies provides genome-wide markers coverage at a low cost per data point, allowing to assess the inheritance of the entire genome with nucleotide-level precision (Cobb et al. 2013, Varshney et al. 2014).
Today, studies are no longer limited by our ability to genotype large populations or by the number of markers, but rather by the high cost and low throughput of phenotypic strategies for traits of interest and in environments relevant to plant breeding (Cobb et al. 2013).
How genomic markers help understand the history of domesticated species?.- Molecular markers that are not affected by selection bring information about demographic histories, including the history of the domestication process (Tang et al. 2010). Earlier models of domestication proposed a single domestication event and suggested that domestication occurred through strong selection and severe genetic bottleneck, which resulted in reproductive isolation between wild and cultivated populations (Meyer & Purugganan 2013). Then, models integrated gene flow and introgression between cultivated and wild relatives, as occurred in grape (Vitis vinifera L. subsp. vinifera), that emerged in Near East region, but evidence of introgression from the wild progenitor (Vitis vinifera subsp. sylvestris) during Europe introduction was found (Myles et al. 2011). High levels of genetic diversity and fast decay of LD in vinifera suggest a weak bottleneck at the beginning of domestication process, followed by thousands of years of vegetative propagation (Myles et al. 2011).
There are some cases in which multiple domestication events occurred. For example, resequencing common bean genomes -- including wild and domesticated forms -- confirmed two independent domestication events, one occurred in Mesoamerica and the other one in the Andean region. Besides, these two gene pools diverged 165,000 years ago, long before domestication started (Schmutz et al. 2014). Finally, an alternative domestication model proposes that crops are domesticated from interspecific hybridization, sometimes followed by clonal propagation. This is common in tree crops, as is the case of species of the genus Citrus, previously discussed. New groups of bananas and plantains developed when diploid domesticated bananas (genome AA) spread into the range of wild Musa balbisiana Colla (genome BB), producing the AAB and ABB triploids (Heslop-Harrison & Schwarzacher 2007).
How is genomic data obtained? Several massive parallel sequencing platforms are commercially available, and while the specific methods vary, they obtain millions of short DNA sequence reads from random locations in the genome (Escalante et al. 2014). Massive sequencing technologies have been widely used for de novo sequencing and several crop and other angiosperm genomes have been sequenced, which are used as reference in no model species studies (Table 1). Some of the genome assemblies are in draft stage and extensive work is ongoing in the direction of closing the gaps and re-sequencing. In addition to the genome sequence, transcriptomes and expressions profiles are also available for many crops. The large genome size and polyploidy exhibited by many crop species make more difficult the sequencing and further analysis. In some allopolyploid crops, the genomes of progenitors species were first sequenced and then were used to assemble the polyploid genomes of the domesticated forms. This strategy was used in cotton (Wang et al. 2012, Li et al. 2014, Li et al. 2015), strawberry (Hirakawa et al. 2014) and peanut (Bertioli et al. 2016).
Table 1 Examples of published sequenced crop genomes. The statistics for each genome are taken from the publication, despite several model plant genomes have had significant updates to genome assemblies and gene counts.
| Species | Common name | Genome size (Mb) | Assembly size | Ploidy and chromosome number | # predicted genes | Reference |
|---|---|---|---|---|---|---|
| Domesticated in Mexico | ||||||
| Amaranthus hypochondriacus | Amaranth | 466 | 377 | 2n = 32 | 23,059 | Clouse et al. 2016 |
| Capsicum annuum | Hot pepper | 3,480 | 3,060 | 2n = 24 | 34,903 | Kim et al. (2014) |
| Carica papaya | Papaya | 372 | 271 | 2n = 18 | 24,746 | Ming et al. (2008) |
| Gossypium hirsitum | Cotton | 2,250-2,430 | 2,173 | 2n = 4x = 52 | 76,943 | Li et al. (2015) |
| Phaseolus vulgaris | Common bean | 587 | 473 | 2n = 22 | 27,197 | Schmutz et al. (2014) |
| Theobroma cacao | Cocoa | 430 | 327 | 2n = 20 | 28,798 | Argout et al. (2011) |
| Zea mays | Maize | 2,300 | 2,048 | 2n = 20 | 32,540 | Schnable et al. (2009) |
| Domesticated in America | ||||||
| Anana comosus | Pineapple | 526 | 382 | 2n = 50 | 27,024 | Ming et al. (2015) |
| Chenopodium quinoa | Quinoa | 1,450-1,500 | 1,390 | 2n = 4x = 36 | 44,776 | Jarvis et al. (2017) |
| Gossypium barbadense | Cotton | 2,470 | 1,395 A subgenome 776 B subgenome |
2n = 4x = 52 | 77,526 | Liu et al. (2015) |
| Manihot esculenta | Cassava | 770 | 532 | 2n = 36 | 30,666 | Prochnik et al. (2012) |
| Nicotina tabacum | Tobacco | 4,500 | 3,700 | 2n = 4x = 48 | 90,000 | Sierro et al. (2014) |
| Solanum lycopersicum | Tomato | 900 | 760 | 2n = 24 | 34,727 | The Tomato Genome Consortium (2012) |
| Solanum tuberosum | Potato | 844 | 727 | 2n = 12 | 39,031 | The Potato Genome Sequencing Consortium (2011) |
| Vaccinium macrocarpon | Cranberry | 470 | 420 | 2n = 12 | 36,364 | Polashock et al. (2014) |
| Other important economic crops | ||||||
| Glycine max | Soybean | 1,115 | 950 | 2n = 20 | 46,430 | Schmutz et al. (2010) |
| Oriza sativa indica | Rice | 466 | 429 | 2n = 24 | 46,022-55,615 | Yu et al. (2002) |
| Oriza sativa japónica | Rice | 420 | 390 | 2n = 24 | 37,544 | Goff et al. (2002) |
| Sorghum bicolor | Sorghum | 730 | 698 | 2n = 20 | 27,640 | Paterson et al. (2009) |
| Triticum aestivum | Bread wheat | 17,000 | 3,800 | 2n = 6x = 42 | 90,000-94,000 | Brenchley et al. (2012) |
Several strategies to construct genomic libraries (collection of DNA that will be sequenced) have been developed. When the genomic library consists in fragments from whole genomes, it is called whole genome resequencing (WGRS). This approach has been used in several crop species, as common bean (Schmutz et al. 2014), rice (Huang et al. 2012, Wang et al. 2016), maize (Hufford et al. 2012, Xu et al. 2014); and soybean (Li et al. 2013) to identify selection signatures and to infer the domestication history. Nevertheless, WGRS is relatively expensive, particularly in large genomes or polyplolid species, reducing the number of samples that can be multiplexed (Kim et al. 2015).
In the last few years, different strategies to genotype a larger number of samples at reduced costs have been developed, including microarrays and methods to construct reduced representation libraries (RRLs). The cost per SNP using microarrays is relatively low but it is necessary to know the sequence of the variants (SNPs) that will be used (Bolger et al. 2014). Construction of RRLs, also known as restriction-associated DNA sequencing (RAD-seq), is a method for sequencing loci adjacent to restriction cut sites across the genome. RAD-seq targets a subset of the genome, therefore allow multiplex a higher number of samples without prior genomic information (Elshire et al. 2011, Andrews et al. 2016). The use of these tools has been intensified to genotype large populations, for both top-down and bottom up approaches. For example, the 539 inbred lines of maize found in the International Maize and Wheat Improvement Center (Centro Internacional de Mejoramiento de Maíz y Trigo, CIMMYT) have been genotyped using genotyping by sequencing (GBS) to know the genetic diversity and structure (Wu et al. 2016).
To design a genomic study it is necessary to consider the size and complexity of the genome, and if a reference genome is available. Besides, the amount of markers that will be identified depends on the approach used to construct the library (WGRS, RAD-seq or microarray). This must be considered within the context of the biological question. For instance, to infer neutral processes, as demographic histories, it only requires from hundreds to a few thousands of molecular markers. If the goal is to identify selection signatures or functionally characterize regions, dozens of thousands to hundreds of thousands of markers are required; for example, for GWAS or gene mapping, at least hundreds of thousands of SNPs are needed (Andrews et al. 2016).
The functional aspect of genomics in domestication studies. The goal of functional genomics is to understand the complex relationships between the genome and the phenotype. This involves dynamic processes as gene transcription, translation, regulation of gene expression and protein interactions. These processes comprise a number of -omics approaches, such as transcriptomics (gene expression), proteomics (protein production), and metabolomics (characterization of metabolic products; Huang et al. 2016). Recently, some studies have added the functional genomics aspect of domestication to assess how many and which genes show differences in expression when wild and domesticated forms are compared.
From the -omics, transcriptomics has received more attention (Huang et al. 2016). The transcriptome is the set of messenger RNA molecules in one cell or a population of cells, and provides information about expression and genic regulation. RNA-seq (RNA sequencing), also called whole transcriptome shotgun sequencing (WTSS), uses massive sequencing to reveal the presence and quantity of RNA in a sample at a given moment in time. RNA-seq is an alternative of genome reduction representation, that can be used to identify differential expressed genes, alternative splicing (mechanism by which different forms of mature mRNAs are generated from the same gene), and genetic regulatory networks, as well as for annotation, and gene and markers discovery (Andrews et al. 2016, Huang et al. 2016). Transcriptomics may complement genomic studies, because RNAseq focused on coding regions, while genomics approaches, as RADseq, include coding and noncoding regions (Andrews et al. 2016).
For example, in developing cotton fiber, ~15 % of 1,300 proteins are significant up or down-regulated (Hu et al. 2013). Most of the changes took place in the early developmental stages, which is consistent with human selection for earlier activation of fiber elongation in domesticated types. Nevertheless, there were a few changes that overlapped between transcripts and proteins, probably because protein abundances depend on multiple factors, as translation, post-translational modifications and degradation processes (Hu et al. 2013).
In maize, Swanson-Wagner et al. (2012) detected hundreds of genes whose expression patterns were altered during domestication, some of them are involved in biotic and abiotic stress responses. More recently, Huang et al. (2016) studied the transcriptomes of six teosinte accessions and found that approximately 75 % of the genes were highly conserved between maize and teosinte. Moreover, they also found 1,516 unigenes (set of transcripts located at the same loci) that were specifically expressed in teosinte, and identified 99 unigenes with strong selection signals, of which 57 might be under strong selection during maize domestication and improvement. This kind of functional studies allows an integrative understanding of the genomic effects that domestications has on species.
Conclusions
The domestication process and breeding have modified the genomes of the plant species that we consume on a daily basis. The recent development of massive parallel sequencing tools allows us to access to information from whole genomes, accelerating identification of genes affected by domestication and to correlate domestication syndrome traits with their molecular basis.
Because many crop species represent a good model for evolutionary studies and have high economic value, there are detailed genetic studies of the most important crops and their wild relatives. From these studies, general patterns have been inferred and in some species, at least partially, the dynamics of the evolutionary process associated to domestication and diversification have been elucidated. Nevertheless, it is necessary to expand our knowledge to no model species, particularly to perennial plants, including tree crops, which have received far less attention. Besides, more functional genomics studies are needed to have an integrative knowledge of the changes that have affected crop species.
Despite major advances of the last years in genomics, important challenges still remain. One important challenge is the analysis of big databases that are generated because computer clusters are frequently needed. Also, the analysis of large, complex and diverse genomes as is the case of conifers is still a relevant problem. We also need to develop efficient approaches to include and analyze repetitive regions. In addition, the identification of paralogues, and the study of polyploid species, remain as some of the most critical problems when analyzing massive sequencing data. Besides, in GWAS, phenotyping is a major operational bottleneck that limits the power and resolution of genetic analysis. There is an urgent need for high throughput, cost-effective, and precise phenotyping methods.
Given the importance of wild progenitors to analyze changes derived from domestication, more attention should be paid to the genetic diversity and adaptation of the wild forms. Besides, in the context of climate change, this approach facilitates the development of climate-tolerant cultivars. In order to achieve these goals, it is necessary to integrate training and research across scientific fields, including genetics, plant breeding, molecular biology, evolution, statistics and bioinformatics.

