











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Study contribution
In this study we characterized whole genome of several influenza A virus isolates obtained from wild birds. The phylogenetic comparison of all individual influenza A viruses segments sequenced in this work showed that they mostly clustered with other isolates from North America, with some being related to viruses circulating in Asia. Our observations suggest that migratory birds, which come to wetlands in Sonora, Mexico, co-migrate along flight corridors with bird populations from different breeding sites in North America, possibly Alaska and Canada, where major flyways may overlap with Asian flyways, allowing the interchange of genetic material and the increase in viral diversity.
Introduction
Migration is one of the important aspects of bird behavior, with billions of specimens migrating thousands of kilometers across the globe. These journeys generally follow a predominantly north to south direction, and link Arctic sites with temperate and tropical areas. Influenza A viruses circulate in many animal species, including humans, pigs, horses, dogs and a wide range of wild and domestic birds.1, 2 The virus reservoir in nature is thought to be formed principally by wild aquatic birds, mainly from Anseriformes (particularly ducks, geese and swan) and Charadriiformes (particularly gulls, terns and waders) orders, which contribute to wide-spread of avian influenza A viruses (IAV) during their seasonal migrations on their migratory routes.2, 3 Eight migratory flyways have been identified globally, with some of them connecting the high arctic north with the Caribbean, up to south Tierra de Fuego, through the Pacific sea coast.2, 4-6 The most important North American flyways identified are Atlantic, Mississippi, Central, and Pacific, where migratory birds stop over wintering grounds along the route. Millions of aquatic birds congregate in their breeding grounds in the northern hemisphere around the world, allowing free transmission of influenza viruses and exchange of viral segments during co-infections. The fact that IAV does not cause disease in wild birds [they are mostly low pathogenic avian influenza, (LPAI)], but are excreted in large quantities in feces, helps their spread. Excreted viruses remain viable during several days, and different viral types have been observed in environmental reservoirs at one point.7 Differences in set of gene segments (assortment of viral segments) among continental population of LPAI viruses have been described, and intercontinental reassortment of viral segments has also been found in migratory birds.4-6, 8, 9 Highly pathogenic avian influenza (HPAI) strains (H5N2, H5N1, H7N3, H9N2, H7N9) circulate widely and were transferred to caged birds.10-11 Recent surveillance study of IAV in aquatic overwintering birds in Guatemala showed a high virus diversity (showing 52 different genomic sets in 68 analyzed isolates), and demonstrated evidence of introduction of a Eurasian viral PA segment into North Americas IAV population.12 In fact, diverse HPAI of Asian-origin have recently been reported in North America.13
There is an increased interest in monitoring the migration and spread of IAV strains through the globe. However, the coverage of wild bird sampling and genetic analysis of IAV is not always complete. For example, data available for Mexico are scarce. A previous eco-epidemiological study demonstrated IAV circulation in migratory birds in the Northwestern Mexican wetlands and partial characterization of hemagglutinin genes of some viruses was performed; however, at that time we could not isolate and characterize whole samples.14, 15 For this reason, in this study we took advantage of high-throughput generation sequencing, to achieve full characterization and perform in-depth phylogenetic analysis of the whole genome of several IAV sampled from wild birds in the Pacific flyway migratory grounds in Mexico.
Materials and methods
Ethical statement
Samples were collected post mortem from birds captured during official surveillance activities conducted by the Mexican Department of Environment and Natural Resouces (Semarnat). At the time of sampling (2007-2009), no further authorization was required.
Collection of samples, RNA extraction and library preparations
Thirteen influenza A virus-positive cloacal swabs, as tested for matrix gene by One-Step qRT-PCR, were collected from opportunistic sampling of 1262 hunter-killed migratory birds as part of a local surveillance program in Sonora, Mexico during the 2007-2008 and 2008-2009 seasons. The collection was realized in Laguna Moroncarit, Huatabampo (26°41'56.4"N 109°31'45.7"W) and EsteroTobari, Cd. Obregon (27°00'43.6"N 109°54'13.1"W).14 Samples were stored at -80oC with minimum handling until extraction (3 and 4 years reaspectively).
Before extracting viral nucleic acids, the samples were incubated with Turbo DNAse (Ambion) to remove external or host DNA. Total RNA for each sample was extracted individually with the PureLink Viral DNA/RNA kit (Invitrogen), using linear acrylamide as a carrier. Individual whole influenza A genome amplification was performed by multi-segment RT-PCR using a set of universal primers, MB-Tuni-12 (5' ACGCGTGATCAGCAAAAGCAGG 3') and MBTuni-13 (5' ACGCGTGAT-CAGTAGAAACAAGG 3') with the SuperScript III one-step RT-PCR platinum Taq HiFi kit (Invitrogen), as described previously.16 All RT-PCR products were purified and concentrated by DNA clean & concentrator kit (Zymo) and visualized in 1.5% agarose gels.
Library preparation and sequencing
PCR products from whole genome RT-PCR were used as input material for preparation of sequencing libraries. DNA was sheared by treatment with NEBNext Fragmentase (New England Biolabs) for 15 min at 37 °C. Sheared DNA was purified and concentrated by DNA clean & concentrator kit (ZYMO) and 50 to 100 ng were used for the preparation of the libraries using Illumina's Genomic DNA Sample Preparation kit with Multiplex Sample Preparation Oligokit, as described by the manufacturer.
High-throughput sequencing was performed at the next-generation sequencing core facility located at the Instituto de Biotecnología, UNAM, in Cuernavaca, Morelos. Libraries of approximately 350 nucleotides were used to generate sequencing clusters, followed by 45 or 66 cycles of single base pair extensions in the Genome Analyzer IIx sequencer (Illumina, San Diego, CA), which was followed by a multiplex barcode acquisition. Computational analysis was performed using the computational cluster at the Instituto de Biotecnología, UNAM. Image analysis was carried out using the Genome Analyzer Pipeline Version 1.4.
All reads were quality filtered, and aligned to the reference genome generated separately for each sample. The number of filtered and influenza specific reads of nine sequenced samples is shown in Table S1. To choose the appropriate reference genome to map the reads, the reads were first mapped to influenza A strains used in Paulin et al.17 and the segments with most of the reads mapped were selected for reference genomes. The alignment was performed by SMALT version 0.7.0.118 and a consensus sequence was called by SAMtools version 0.1.18. To allow for sequence variability, two separate alignment rounds were performed. In the first round, 15 and 20 mismatches were allowed with 46 or 65 nucleotide long reads, respectively, while during the second alignment round only 5 mismatches were allowed using as a reference the consensus sequence previously generated during the first mapping round. Reads that did not match the reference genome were not considered for further analysis.
Phylogenetic analysis
Phylogenetic analysis was performed separately for each influenza A segment, and in the case of the HA and NA segments, for each identified subtype. Included sequences were selected from four regions reflecting continents where avian species used in this study are observed: Europe (EU), North America (NA), South America (SA), and Asia (AS). Sequences from avian, human, equine and swine sources were selected from NCBI Influenza Virus Resource, and ones with identical nucleotide coding sequences were collapsed. Additionally, sequences with more than 20 consecutive N's were eliminated.
The resulting sequences were clustered into arbitrary groups for each segment/subtype to remove highly similar (but not identical) sequences with cd-hit.19 For the case of the hemagglutinin and neuraminidase, all the clusters were done at 95 % of sequence identity for a median of 54 sequences per subtype for all samples included, with two outliers: N1 and N2 with 178 and 298 sequences, respectively.
For the rest of the segments, the clusters were constructed at 92 % of sequence identity, having a median of 90 sequences per tree (min = 54, mean = 85, max = 104). The number of sequences used is shown in Table S2, For the selected sequences, the name was changed as follows: the continent of origin, abbreviated source, accession number, gi, species of origin, virus name and number, year, segment number, segment name and for the case of hemagglutinin and neuraminidase, the subtype as shown in the following example: NA_AV_GU051078_269822049_A_semipalmatedsandpiper_Delaware_2144_2000_3_PA. Finally, each field was separated by an underscore instead of a slash to avoid issues with the software used.
Each data set was aligned in nucleotides with the multiple sequence comparison by log-expectation (MUSCLE) algorithm20 using the default parameters. The presence of mismatches or possible frame shifts was checked after translation to protein. The substitution model was calculated for each alignment with IQTree, in model mode.21 The phylogenetic trees were constructed with the IQTree algorithm, using the model selected in the previous step, using the ultrafast bootstrap option with 10 000 bootstraps.22 In the phylogenetic trees, only the geographical area is shown. The strains sequenced in this study are marked with a black dot next to their name. The bootstrap value for each split is shown in a circle, which the color intensity code indicates its value as a percentage.
The isolates sequenced in this work were grouped into clades after visual inspection of phylogenetic trees. The clades are well defined and the split that separates them has an ultra-fast bootstrap close to 100 percent.
Results and discussion
In total, thirteen avian influenza-positive samples were obtained from 7 green winged teals (Anas crecca carolinensis), 1 gadwall (Anas strepera), 1 American wigeon (Anas americana), 3 northern shovelers (Anas clypeata) and 1 redhead (Aythya americana). Samples were collected from two migratory avian wintering grounds in Sonora, Mexico; 3 were collected during 2007-2008 wintering season and 10 from the 2008-2009 season.14 Both locations (Laguna Moroncarit, Huatabampo and Estero Tobari, Cd. Obregon) are among the most important wetlands for avian migration on the Mexican Pacific coast. Given that in previous studies only partial identification of genes was achieved,14, 15 high-throughput sequencing was used to improve our data. Using of high-throughput sequencing technologies has shown usefulness for avian influenza virus complete genomic characterization from field samples, otherwise limited using traditional virological approaches.23
A whole genome sequence with >100x coverage was obtained from 7 samples, which were used in the following analysis, while the remaining six samples generated only partial sequences, allowing only a description of influenza virus subtypes (Table 1). The samples from 2007-2008 season were identified as H6N1 (A/Green-winged teal/Mexico-Sonora/266/2008), and H5N2 (A/Gadwall/Mexico-Sonora/150/2008; A/Redhead/Mexico-Sonora/408/2008) (Table 1).
Table 1 Avian influenza A from waterfowl analyzed in this study
| Sample | Species | Sex | Date of collection |
WG1 | Subtype2 |
|---|---|---|---|---|---|
| A/gadwall/Sonora/150/07 | Gadwall | F | 15/12/2007 | ND3 | H5N2 |
| A/green winged teal/Sonora/266/08 | Green winged teal | M | 19/01/2008 | H6N1 | H6 |
| A/redhead/Sonora/408/08 | Redhead | M | 12/02/2008 | ND | H5N2 |
| A/green winged teal/Sonora/701/08 | Green winged teal | F | 13/12/2008 | H11N3 | |
| A/northern shoveler/Sonora/738/08 | Northern Shoveler | M | 13/12/2008 | H6N1 | H6 |
| A/American wigeon/Sonora/769/08 | American Wigeon | F | 14/12/2008 | H9N2 | H9 |
| A/northern shoveler/Sonora/796/08 | Northern Shoveler | F | 14/12/2008 | ND | H5N3 |
| A/northern shoveler/Sonora/797/08 | Northern Shoveler | F | 14/12/2008 | H5N3 | H5 |
| A/green winged teal/Sonora/827/09 | Green winged teal | F | 10/01/2009 | ND | H5N3 |
| A/green winged teal/Sonora/829/09 | Green winged teal | F | 10/01/2009 | H6N5 | |
| A/green winged teal/Sonora/840/09 | Green winged teal | F | 10/01/2009 | ND | H5N3 |
| A/green winged teal/Sonora/1116/09 | Green winged teal | M | 08/02/2009 | H10N7 | |
| A/green winged teal/Sonora/1132/09 | Green winged teal | M | 08/02/2009 | ND | H10N3 |
1 WG. The whole genome was determined in this study.
2 Subtype as determined previously (Montalvo-Corral and Hernandez 2010) or in this study.
3 Not determine the whole genome.
The samples from the 2008-2009 season were identified as H5N3 (A/Northern shoveler/Mexico-Sonora/796/2008; A/Northern shoveler/Mexico-Sonora/797/2008); A/Green winged teal/Mexico-Sonora/827/2008; A/Green winged teal/Mexico-Sonora/840/2008), H6N1 (A/Northern shoveler/Mexico-Sonora/738/ 2008), H6N5 (A/Green winged teal/Mexico-Sonora/829/2009), H9N2 (A/American wigeon/Mexico-Sonora/769/2008), H10N3 (A/Green winged teal/Mexico-Sonora/ 1132/2009), H10N7 (A/Green winged teal/Mexico-Sonora/1116/2009) and H11N3 (A/Green winged teal/Mexico-Sonora/701/2008).
It is of interest that out of the 7 isolates characterized from green winged teal; six different hemagglutinin and neuraminidase combinations were found (H5N3, H6N1, H6N5, H10N3, H10N7, H11N3), with only H6N1 being shared by two birds (Table 1). The H5N3 subtype was found in two Northern shovelers, collected at Estero Tobari, and in Laguna Moroncarit in two Green winged teals. The H5N2 combination was also found more than once, however, in different avian species (Table 1).
Whole genome sequencing allows analysis of all influenza segments, which could have important implications in influenza pathogenesis. While PB2 and PB1 gene segments from the field samples sequenced in this work clustered together, mostly with other viruses isolated from North American territories (Figures S1 and S2), the PA gene segments clustered into two separate groups (Figure 1), with both groups localized close to the North American isolates. Phylogenetic analysis of genomic segments corresponding to nucleoprotein, matrix and nonstructural proteins showed similar results as those observed for the polymerase gene segments. While matrix genes formed one group, close to other samples from North America (Figure S3), one Mexican sample (A/Green winged teal/Mexico-Sonora/1116/2009 H10N7) grouped separately in its NS gene segment (Figure 2), and two samples, (A/American wigeon/Mexico-Sonora/769/2008 H9N2; A/Green winged teal/Mexico-Sonora/1116/2009 H10N7) grouped separately from the remaining Mexican samples in the NP gene segment (Figure 3). NS segment, which groups separately, is closely related to other North American samples, while the NP segments were close to isolates from Asia. These observations confirm the complexity of influenza virus inter-continental circulation.
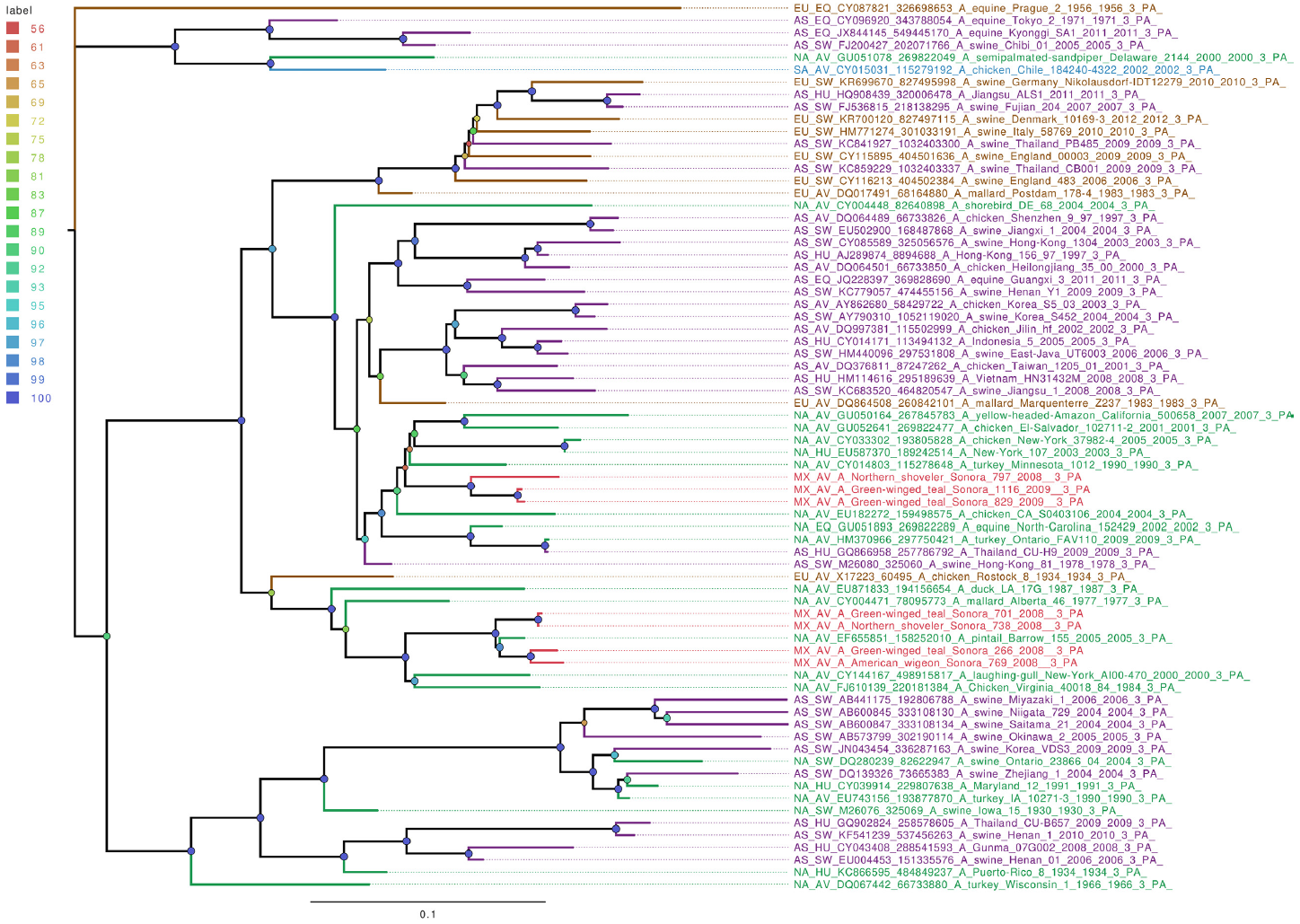
Figure 1 The phylogenetic tree for the PA gene segment of avian influenza viruses was analyzed in this work. For used sequences, the name was changed as follows: continent of origin, source, gi and accession number, virus name, segment number and segment name. In phylogenetic trees, following area code was used: Europe (EU), North America (NA), South America (SA), Asia (AS) and Mexico-this work (MX). The bootstrap value for each split is shown in a circle with the intensity color code indicating its value as a percentage. The color code of the continent of origin is as follows: green North America (NA), blue South America (SA), brown Europe (EU) purple Asia (AS), red sample sequences in this work (MX).
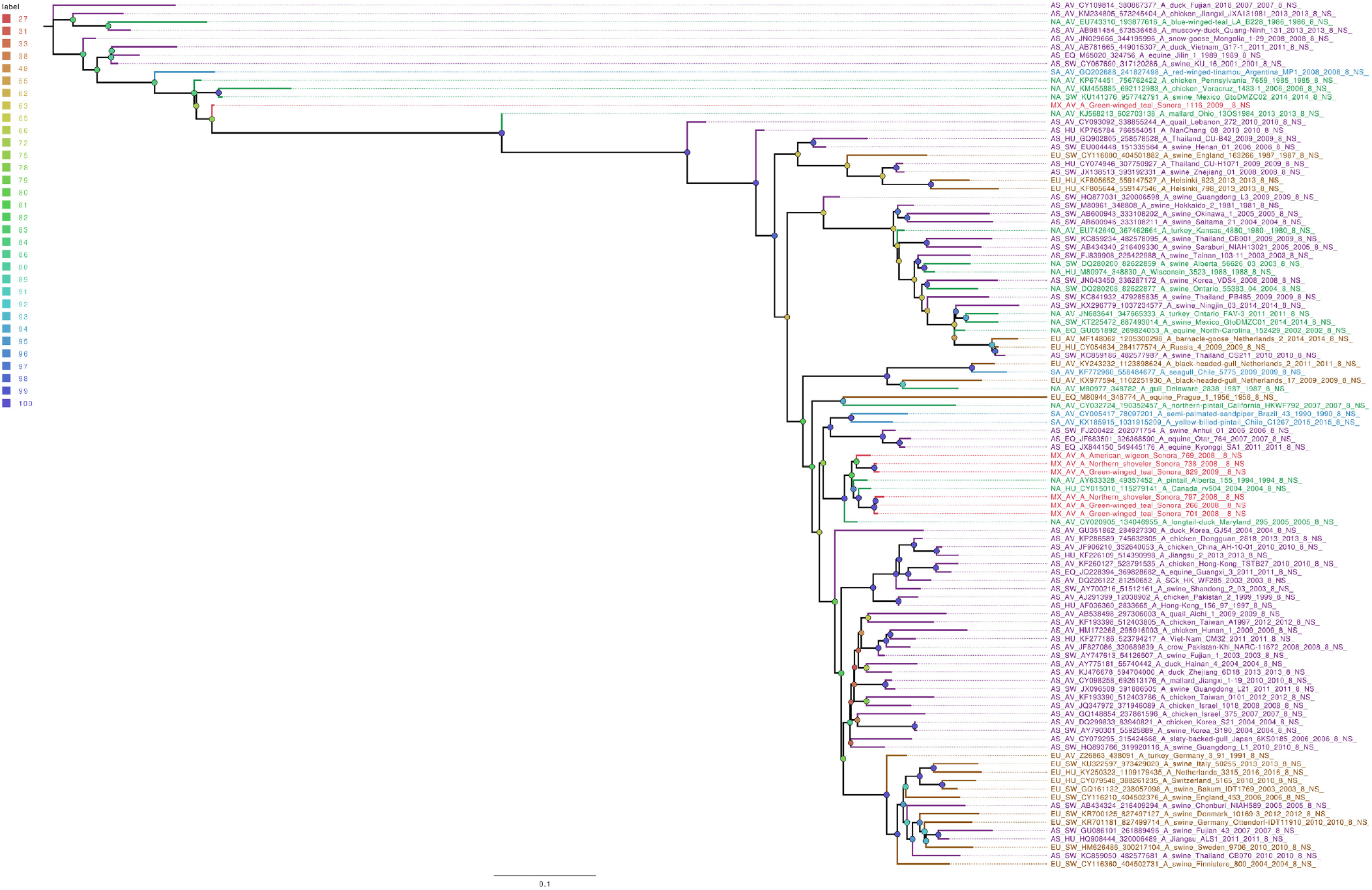
Figure 2 The phylogenetic tree for the NS gene segment of avian influenza viruses was analyzed in this work. For the selected sequences, the name was changed as follows: continent of origin, source, gi and accession number, virus name, segment number and segment name. In phylogenetic trees, following area code was used: Europe (EU), North America (NA), South America (SA), Asia (AS) and Mexico-this work (MX). The bootstrap value for each split is shown in a circle with the intensity color code indicating its value as a percentage. The color code of the continent of origin is as follows: green North America (NA), blue South America (SA), brown Europe (EU) purple Asia (AS), red sample sequences in this work (MX).
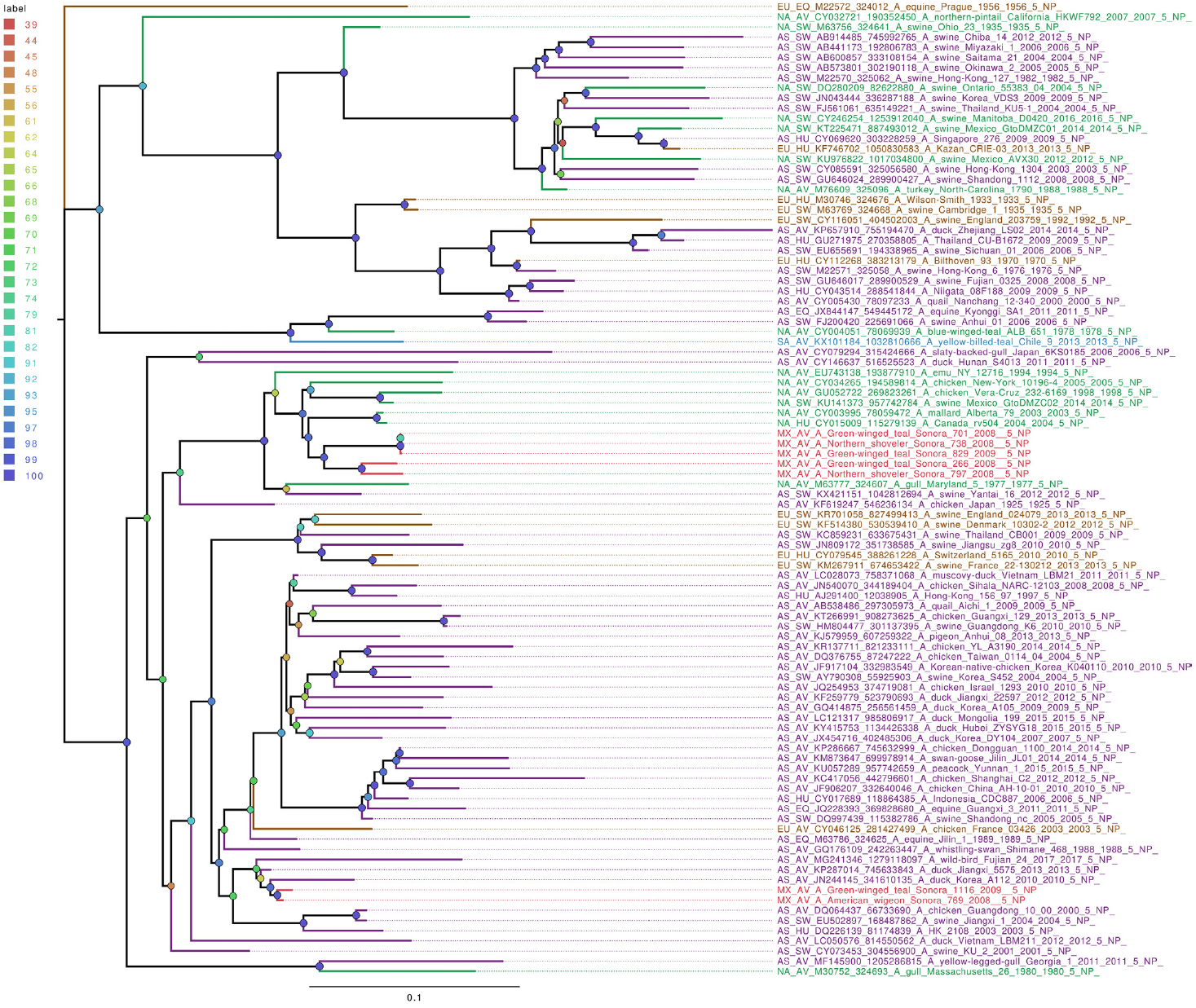
Figure 3 The phylogenetic tree for the NP gene segment of avian influenza viruses was analyzed in this work. For used sequences the name was changed as follows: continent of origin, source, gi and accession number, virus name, segment number, segment name and for the case of hemagglutinin and neuraminidase the subtype. In phylogenetic trees, following area code was used: Europe (EU), North America (NA), South America (SA), Asia (AS) and Mexico-this work (MX). The bootstrap value for each split is shown in a circle with the intensity color code indicating its value as a percentage. The color code of the continent of origin is as follows: green North America (NA), blue South America (SA), brown Europe (EU) purple Asia (AS), red sample sequences in this work (MX).
Hemagglutinin and neuraminidase gene segments
From the seven wild bird IAV-positive samples sequenced, five different hemagglutinin types were found (H5, H6, H9, H10, and H11), with the HA6 type being the only present more than once. Phylogenetic analyses of hemagglutinin gene segments showed that they are closely related to other IAV from North America (Figures S4-S6). The exceptions were H6 and H11 segments, which were grouped closer to isolates from Asia (Figure 4 and 5). Interestingly, the 3 Mexican HA6 samples found in this study were grouped together despite being isolated in two different influenza seasons, from two distinct locations. Sample A/Green-winged teal/Mexico-Sonora/266/2008 H6N1, was collected during season 2007-2008, in Estero Tobari, and samples A/Northern shoveler/Mexico-Sonora/738/2008 H6N1, and A/Green-winged teal/Mexico-Sonora/829/2009 H6N5, were collected during 2008-2009 season, the first from Estero Tobari and the second sample from Laguna Moroncarit (Figure 4). However, sample A/Green winged teal/Mexico-Sonora/701/2008 localized close to the Asian sample described in 2016.

Figure 4 The phylogenetic tree for the H6 gene segment of avian influenza viruses was analyzed in this work. For used sequences the name was changed as follows: continent of origin, source, gi and accession number, virus name, segment number, segment name and for the case of hemagglutinin and neuraminidase the subtype. In phylogenetic trees, following area code was used: Europe (EU), North America (NA), South America (SA), Asia (AS) and Mexico-this work (MX). The bootstrap value for each split is shown in a circle with the intensity color code indicating its value as a percentage. The color code of the continent of origin is as follows: green North America (NA), blue South America (SA), brown Europe (EU) purple Asia (AS), red sample sequences in this work (MX).
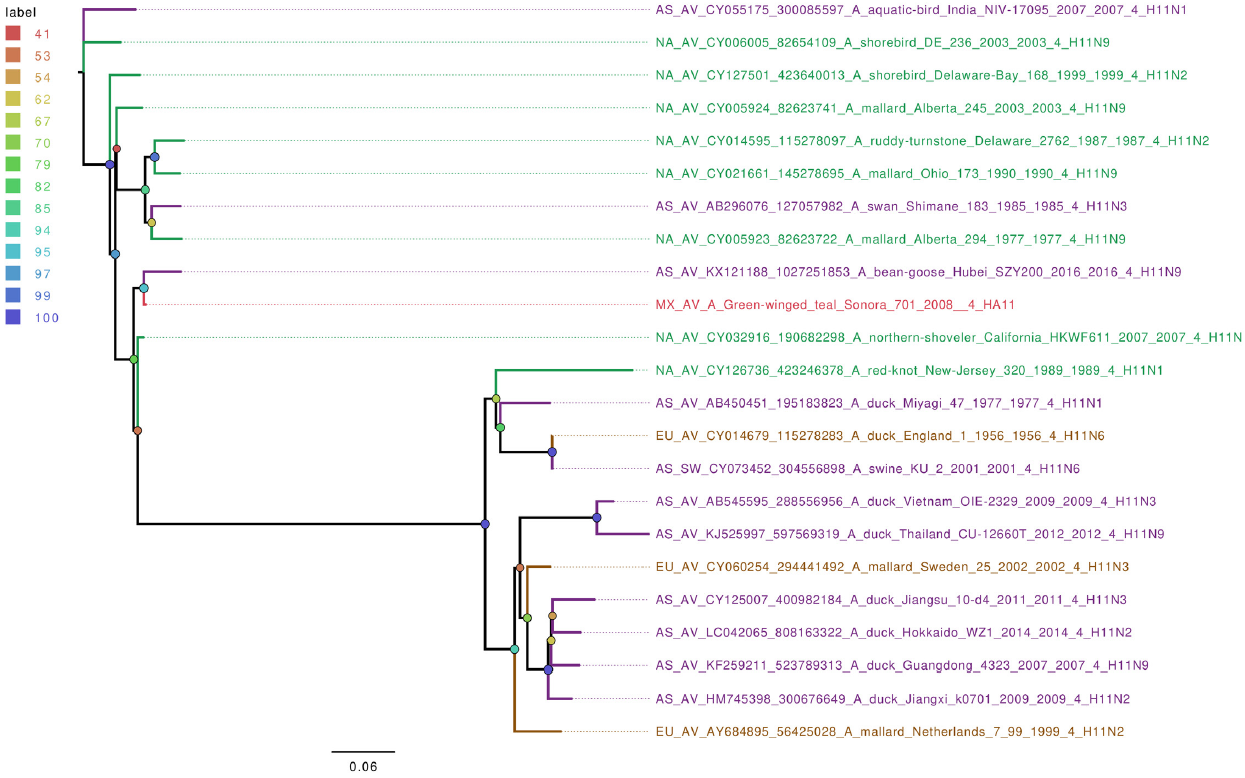
Figure 5 The phylogenetic tree for the H11 gene segment of avian influenza viruses was analyzed in this work. For used sequences the name was changed as follows: continent of origin, source, gi and accession number, virus name, segment number, segment name and for the case of hemagglutinin and neuraminidase the subtype. In phylogenetic trees, following area code was used: Europe (EU), North America (NA), South America (SA), Asia (AS) and Mexico-this work (MX). The bootstrap value for each split is shown in a circle with the intensity color code indicating its value as a percentage. The color code of the continent of origin is as follows: green North America (NA), blue South America (SA), brown Europe (EU) purple Asia (AS), red sample sequences in this work (MX).
Analysis of the H5 segment characterized in this work showed that sample A/ Northern shoveler/Mexico-Sonora/797/2008 H5N3, separated from the previously characterized Mexican H5 samples isolated in 1994-1997 period (Figure S4). This genetic distance might be explained by differences in space-time genetic changes and possible origins, because samples from 1994-1997 were isolated from farm chickens and belonged to an HPAI virus. AIV H5 subtype characterized in this work is low pathogenic avian influenza (LPAI) virus, lacks of hemagglutinin pathogenic markers. The occurrence of the H5 subtype is not as rare as previously thought in wild birds. A large-scale surveillance conducted in the USA between 2006 and 2011 found H5 subtype highly represented among IAV isolates24 and HPAIV H5 was recently found in wild birds in Canada and USA.25 In our study, which included few samples, 6 of 13 IAVs were subtyped as H5 isolates (Table 1). Alternatively, recent data from ducks overwintering in Guatemala showed that only 3 samples from 68 characterized isolates were H5.12
With respect to neuraminidase genome segments, five different NA types were identified (N1, N2, N3, N5, and N7), with N1 and N3 types being present in two samples (Table 1), and all are closely related to other North American isolates (Figures S7-S11).
To analyze virus diversity, specific combinations of viral segments in each sample were determined. For this, each segment was classified within the corresponding clade as described in the Materials and Methods. Among the seven fully sequenced isolates, 4 different sets of genome segments were observed (Table 2). First is formed by three viruses (A/greenwingedteal/Sonora/266/08, A/greenwingedteal/Sonora/701/08, and A/northernshoveler/Sonora/738/08), which have all segments in main groups, the second constellation contained two viruses (A/northern-shoveler/Sonora/797/08 and A/greenwingedteal/Sonora/829/09), which differ in 116 nucleotides in the PA segment, with the last two having just one example, A/american wigeon/Sonora/769/08, with the NP segment different from the rest, and lastly most divergent sample A/green winged teal/Sonora/1116/09, with PA, NP and NS segments distinct (Table 2). When H and N genes were added for comparison, 6 different sets of genome segments are observed (Table 2).
Table 2 Sets of genomic segments of avian influenza viruses identified in waterfowl species from Pacific wetlands
| Sample | PB2 | PB1 | PA | NP | M | NS | HANA |
|---|---|---|---|---|---|---|---|
| A/green winged teal/Sonora/266/08 | 1 | 1 | 1 | 1 | 1 | 1 | H6N1 |
| A/green winged teal/Sonora/701/08 | 1 | 1 | 1 | 1 | 1 | 1 | H11N3 |
| A/northern shoveler/Sonora/738/08 | 1 | 1 | 1 | 1 | 1 | 1 | H6N1 |
| A/american wigeon/Sonora/769/08 | 1 | 1 | 1 | 2 | 1 | 1 | H9N2 |
| A/northern shoveler/Sonora/797/08 | 1 | 1 | 2 | 1 | 1 | 1 | H5N3 |
| A/green winged teal/Sonora/829/09 | 1 | 1 | 2 | 1 | 1 | 1 | H6N5 |
| A/green winged teal/Sonora/1116/09 | 1 | 1 | 2 | 2 | 1 | 2 | H10N7 |
When analyzing all 13 IAV isolates characterized (seven complete whole genomes and six partial genomes that identify the HA and NA subtypes), eight different HA and NA combinations were found, confirming the subtype diversity present in the North American Pacific flyway. This is true especially considering that we could analyze samples from only five different avian species. These results again suggest that there is diversity of influenza viruses present in the wintering sites analyzed. Similar high genomic variability was observed in Guatemala, where out of 68 influenza isolates characterized, 19 different subtype combinations were found (nine HA subtypes and seven NA subtypes).12 The Mississippi migratory flyway also showed a high level of genomic variability, with 46 unique HA and NA combinations observed in 297 isolates.26 However, in Kazakhstan, lower variability of HA and NA combinations was observed during a 7 year span (from 2002 to 2009).27
Despite the large dataset of influenza genome sequences now available, knowledge of complete avian influenza virus sequences from wild birds from Mexico and Latin America are very limited.28 Thus, the phylogenetic comparison of all individual IAV segments sequenced in this work showed that they mostly clustered with other isolates from North America. The only exceptions were some of the segments of samples A/Green-winged teal/Mexico-Sonora/266/2008); A/Northern shoveler/Mexico-Sonora/738/2008; and A/Green winged teal/Mexico-Sonora/829/2009 (H6 gene), A/Green winged teal/Mexico-Sonora/701/2008 (H11 segment) and A/American wigeon/Mexico-Sonora/769/2008 and A/Green winged teal/Mexico-Sonora/1116/2009 (NP genomic segment), which appear related to viruses circulating in Asia. These observations support our previous data and show new findings, suggesting that migratory birds, which come to wetlands in Sonora, Mexico, co-migrate along flight corridors with bird populations from different breeding and stop over sites in northern locations of North America, possibly Alaska and Canada, where major flyways may overlap with Asian flyways, allowing the interchange of genetic material and the increase of viral diversity.
Conclusions
Previously we have described isolation and partial characterization of influenza virus isolated from migratory birds in two wetlands in Mexico. In this work using whole genome sequencing we have identified eight different HA and NA combinations in thirteen analyzed samples, while the analysis of seven whole genome samples demonstrated four different combinations of viral segments, suggesting a diversity of circulating IAV. Phylogenetic analysis of samples whose whole genomes were sequenced showed that they are all LPAIV and closely related to North American IAV, with some segments (NP, H6 and H11) being related to strains circulating in Asia. The use of high-throughput technologies enabled genetic characterization of field samples and contributed to fostering our knowledge of IAV genomics and virus circulation in Mexico, a lowly surveyed but biological and epidemiological relevant region, and highlights the need for future studies in Mexican wetlands.
Data availability
Sequences are publicly available at the National Center for Biotechnology Information (NCBI: https://www.ncbi.nlm.nih.gov), accession numbers KY575169-KY575224, and KY593192-KY593208.
Funding statement
This work was supported by grant PICOSI09-209 from the Instituto de Ciencia y Tecnología del Distrito Federal, México, awarded to PI, and by grant IN213410 from UNAM, DGAPA (https://dgapa.unam.mx/) awarded to PI. LFP was supported by a scholarship from CONACYT-Mexico (https://conacyt.mx/).
Conflicts of interest
The authors have no conflict of interest to declare in regard to this publication.
Author contributions
Conceptualization: P Iša
Methodology: P Iša, LF Paulin
Supervision: S López, CF Arias
Formal analysis: P Iša, LF Paulin
Funding acquisition: P Isa
Resources: J Hernandez, M Montalvo-Corral
Writing - original draft: P Iša, LF Paulin, M Montalvo-Corral
Writing - review and editing: P Iša, LF Paulin, J Hernandez, M Montalvo-Corral, S López, CF Arias

